Previous Issues Volume 8, Issue 1 - 2024
Synthesis and Antidiseas Evaluation of Some Bis-Heteroaryls (Thiophene or Selenophene) 1,2,3-triazoles C-trimethylsilylated
Paolo Zanirato1,*, Silvia Businelli2
1Academy of Sciences of Bologna, via Zamboni 31, Bologna, Italy
2Fondatrice Mirasolvia, Italy
*Corresponding author: Dr. Paolo Zanirato, Academy of Sciences of Bologna, via Zamboni 31, Bologna, Italy, E-mail: [email protected]
Received Date: June 07, 2024
Published Date: June 28, 2024
Citation: Zanirato P, et al. (2024). Synthesis and Antidiseas Evaluation of Some Bis-Heteroaryls (Thiophene or Selenophene) 1,2,3-triazoles C-trimethylsilylated. Mathews J Pharma Sci. 8(1):29.
Copyrights: Zanirato P, et al © (2024).
ABSTRACT
This experimental work is part of a research project that includes both the aspect relating to the synthesis, by the 1,3-dipolar cycloadditions (1,3-DC), of a series of new compounds, 1-heteroaryl-1,2,3-triazoles C-4 (or C-5) silylates, and the study of their potential biological applications [1].
The eleven triazole compounds, obtained by reaction of a heteroaryl azide (1a-11a) and trimethylsily acetylene (TMSiAc), directly provides the corresponding heteroaryl triazole ring, at room or low temperature, by 1,3-DC occurring regioselectively with high yelds [2-4].
The discussion is therefore into three parts; in the first part the topic relating to the synthetic procedure for each of the compounds (1-11) is specified. Consequently, this part includes the study regarding the electronic structure of the heteroaryl azido group and the 1,3-dipolar cycloadditions (1,3-DC’s) reaction leading to the final triazole adduct [5-8].
The second part deals with the characterization and structural study of the synthesized (azides and triazoles) products through IR, 1H- and 13C-NMR and mass spectroscopic analysis (MS).
The third part is aimed at determining the potential pharmacological activity of the newly synthesized triazoles. The products were analyzed using appropriate test, from the experimental protocol validated by specialized American research institutes such as the Tuberculosis Antimicrobial Acquisition Facility (TAACF) and the National Cancer Institute (NCI) [9,10].
Keywords: Heteroaryl Azides, 1,3-dipolar Cycloadditions, Heteroaryl Triazoles, C4-triazoles Silylated, Spectroscopic Analysis, Tubercolosis, Cancer.
INTRODUCTION
The constant need to discover new bio-modulators protected by greater activity, great specificity and reduced toxicity, has driven scientific research, both in the medical and agricultural fields, towards the use of a growing number of molecules containing the 1,2,3-triazole system. Since the 1978 our laboratory has produced these compounds by 1,3-dipolar cycloadditions of a heteroarylazide with a diene or a diyne [11-15]. Ultimately, given the urgent need for the development of new anti-virus and anti-cancer drugs, the pharmaceutical industry is making a significant effort to diversify research work, and the future of the industry depends mainly from the realization of this objective. Therefore, currently, many biologically active drugs are either totally synthetic (xenobiotic) or semi-synthetic molecules and a significant part of them is obtained by the incorporation of heteroaryl-triazole systems.
In the last twenty years, the European pharmaceutical industries found their best researchers coming from the world of biology, leading to the so-called "crisis of chemists" who saw their prestige in the industrial field diminish. Now with the advent of combinatorial chemistry, “Chemistry is undoubtedly essential,” says Paul Herrling, research director at Novartis Pharma [16] and molecular biochemists and geneticists, who have bioinformatics knowledge, are the essence of it.
The importance and need for close multidisciplinary collaboration are in fact clearly visible in the research and development of a new drug, where synthetic chemical research is followed by preclinical and clinical testing. Responsibility for the phases that constitute the entire process is entrusted respectively to researchers with chemical-synthetic, bio-toxicological experience and to personnel qualified doctor.
RESULT AND DISCUSSION
It would be useful to have a general 1,3-DC method based on a single dipolarophile alkyne that acts as a solvent/co-reagent at room temperature with a wide number of heteroaryl azides . We carried out here 1,3-DC studies using excess trimethylsilyl acetylene (TMSAc) as a dipolarophile with high steric hindrance and a series of heteroarylaryl azides (1a-11a).
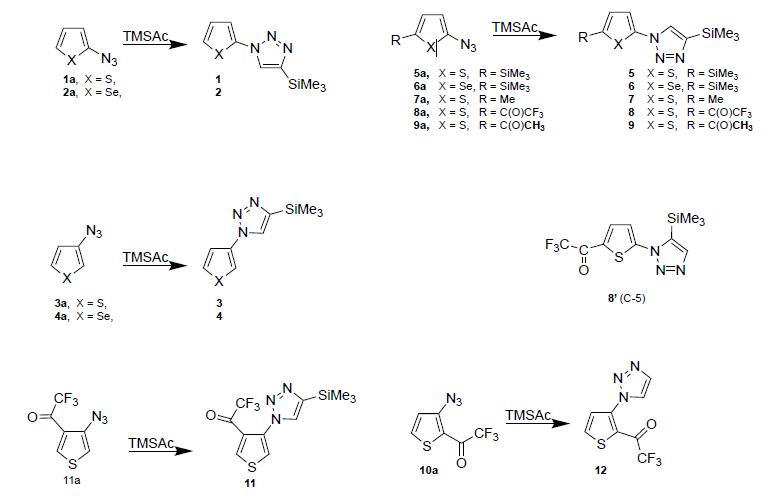
Scheme 1. 1.3-Dipolar cycloaddition reactions of heteroaryl azides 1a-11a with TMSAc.
- 2-azido, (1a) and 3-azido-thiophene (3a),
- 2-azido, (2a), and 3-azido-selenophene (4a),
- 5-trimethylsilyl-2-azido thiophene(5a),
- 5-trimethylsilyl-2-azido selenophene (6a),
- 5-methyl-2-azido thiophene (7a),
- 5-trifluoroacetyl-2-azido thiophene (8a),
- 5-acetyl-2-azido thiophene (9a)
- 2-trifluorocetyl-3-azido thiophene (10a).
- 4-trifluorocetyl-3-azido thiophene (11a),
The standard reactions are classified by the nature of the azido group and indicate that the heteroaryl azides carrying conjugated strong electron-withdrawing groups react about six times faster than those carrying electron-donating groups, furthermore the polarity of the azido group appears to be slightly inverted [17].
Scheme 1. X = S, Se; R = H, Me, SiMe3, С(О)СН3, C(O)CF3.
It must be emphasized once again that the TMSiAc 1,3-DCs carried out at low to room temperatures can be considered a useful test of azido stability-reactivity, especially for the unstable 2-azidoheteroaryls. The reactions occurs almost quick without the presence of a metal catalyst [5a] or high pressure assisted and the additions are generally regiospecific giving C-4’ trimethylsilyl 1,2,3-triazoles as the main products in the case of unstable 2-azidoheteroaryl derivatives, such as 2-azido-selenophene 2a (60%), 2-azidothiophene 1a (whereas more stable starting azides give C-4’substituted triazoles in higher yields, for example 3-azidothiophene (3a, 90%) [17,18].
The ease and useful reaction with TMSiAc to 1,2,3-triazoles deserves some consideration: all of the 2-azido- and 3-azido-heteroaryl previously prepared are used for 1,3-DCs in the presence of TMSAc, as a possible source of biheteroaryls C-4’ trimethysilyl triazoles, whit the exception of the azides 8a and 10a were observed [19-21].
A theoretical study, based on frontier molecular orbital/Pearson’s hard–soft acid–base (FMO/HSAB) principles was applied for the high regiospecificity of the resulting 1-heteroaryl 1,2,3-triazolines (1-9) [22].
Experimental section
The ‘azido transfer’ synthetic protocol offers virtually limitless opportunities for the synthesis of azides derived from heteroaryl systems containing one or more heteroatoms that can undergo regioselective lithiation. This opinion is confirmed by the series of azido-compounds prepared up to now by processes that are essentially based on the ‘azido transfer’ protocol [23,24]. We extended the slightly modified Smith protocol to the syntheses of the eleven azido hetroaryls (1a-11a) obtained from corresponding lithium derivatives, by reaction with tosyl azide.
Materials
Tosyl azide [25] N,N-dimethyl 2,2,2-trifluoroacetamide (N,N-DMTA) 2,3-dibromo- and 3,4 dibromo-thiophene were prepared in accordance with procedures described in the literature [26-28]. Thiophene, 2,5-dibromothiophene, butyl-lithium (1.6 M, hexane solution), N,N-dimethylacetamide (N,N-DMA) and trifluoroacetic anhydride were purchased from Aldrich Chimica Italiana.
Spectra
The IR spectra were recorded with a Perkin-Elmer Model 257 instrument. The 1H- and 13C-NMR spectroscopic data were obtained with a Varian Gemini 200 (or 300) MHz instrument in solution; the values of the J coupling constants are given in Hz. A tool has been used VG Analitycal 7070E to record mass spectra (electronic impact) and an HP 5970 GC/MS instrument for mass-gas chromatography.
The computational calculations were made at the MNDO-AM1 level (PM3 for selenophene derivatives) using the MacSpartan Plus application operating on a Power Macintosh 9100/300Hz and a graphical interface to draw and view all structures. Geometries have been optimized to the minimum of energy using the PRECISE convergence criterion and the options set. The geometries of each individual compound are reported in the corresponding structures together with the significant geometric parameters, distribution of charges, energies and dipole moments.
General procedure for the preparation of 2-azidoheteroaryls (1a, 2a) and 2-azido-5-trimethylsilyl thiophene (5a), 2-azido-5-trimethylsilyl selenophene (6a) and 2-azido-5-methylthiophene 7a
A solution of the appropriate heteroarylbromide (0.08 mol) in anhydrous diethyl ether (100 cm3) was added, at room temperature, under stirring and in nitrogen current, with butyl-lithium in n-hexane (1.6 M; 50 cm3). The resulting reaction mixture was heated to reflux for one hour, then cooled to -70 °C and added drop by drop with a solution of tosyl azide (0.08 mol) in anhydrous ether (100 cm3). Once the adding is completed, the reaction is continued under agitation at 0 °C for 5 h. The pale yellow triazene salt thus obtained was quickly filtered and suspended in pentane. The suspension is treated at 0 °C with a solution of tetrasodium pyrophosphate (0.08 mol) in water (200 ml). The organic layer of pentan is separated and anhydrified on sodium sulfate; by elimination under vacuum ('Rotavapor') of the excess solvent is obtained a residue that has been chromatographed on a column of 'Florisil' using pentane as an eluent. For the individual azides the following yields were obtained: 1a (0.044 mol, 55%), 2a (0.027 mol, 33%), 5a and 6a (0.055 mol, 69%), 7a (0.042 mol, 52.5%) as unstable light yellow oils The para-like substituent effect on fragmentation of the azido group [29,30].
Preparation of 3-azido thiophene (3a) and 3-azido selenophene (4a)
A solution of the appropriate 3-bromoheteroaryl (0.08 mol) in anhydrous diethyl ether (100 cm3) was added under stirring and in nitrogen current at -70 °C with butyl-lithium (solution in hexane 1.6 M, 50 cm3 for compound 2a and solution in cyclohexane 2.2 M, 36 cm3 for 4a). The reaction mixture was stirred for a further 45 minutes (10 min for 4) after which a solution of tosyl azide (0.08 mol) in anhydrous ether (100 cm3) was added drop by drop. Once the addition is completed, the resulting mixture is left under stirring at 0 °C for 5 h. The pale yellow triazene salt thus formed was quickly filtered and suspended in pentane. The suspension was treated at 0 °C with a solution of sodium pyrophosphate (0.08 mol) in water (200 ml) to eliminate the azido group. The light yellow layer of pentane containing the azide derivative was collected and the excess solvent was removed under vacuum. An oily residue is obtained that has been chromatographed on a column of 'Florisil' using pentane as an eluent. For individual azides the following yields were obtained: 3-azidothiophene 3a (0.044 mol, 55%; e.g. 55-56 °C/15mm) and 3-azidoselenophene 4a (0.023 mol, 29%) [31,32].
Preparation of 5-(trifluoroacetyl)-2-azido thiophene (8a), 5-(acetyl)-2-azido thiophene (9a), 2-(trifluoroacetyl)-3-azido thiophene (10a) and 4-(trifluoroacetyl)-3-azido thiophene (11a)
The reactions were performed according to the ‘one-pot’ procedure entailing stepwise halogen-lithium exchange to the corresponding dibromothiophene with equivalent aliquots of butyl-lithium and in turn the resulting thenyl-lithium with N, N-dialkylamides (N,N-dimethylacetamide and N,N-1,1,1-trifluoroacetamide) and tosyl azide [33]. Regioselectivity was achieved by metal-halogen exchange using 2,5-dibromo thiophene (for compound 8a and 9a), 2,3-dibromo thiophene (10a) and 3,4-dibromo-thiophene (11a).
Reactions of the heteroaryl azides 1a-11a with trimethylsilyl acetylene. General procedure
A solution of the appropriate azido-thiophene 1a-11a in pure trimethylsilylacetylene (0.7 M, 3 cm3) was reacted in a closed tube at room temperature (except 1a, 2a and 7a at +4-5 degrees) and in the dark, until a TLC analysis showed the absence of the starting azide. The residue, obtained after careful elimination of excess solvent/reagent at reduced pressure, was analyzed for 'H-NMR and purified by silica column chromatography using hexane as eluent with increasing aliquots of diethyl ether (up to 80%).
1-(2-Thiophene)-4-(trimethylsilyl)-1,2,3-triazole (1)
(5 days, 75%), m.p. 72-74°C;
vmax/cm-1 3140, 2960, 1250, 850 (SiMe3) and 760;
δH (200 MHz; ) 7.82 (1 H, s, H-5), 7.19 (1 H, dd, J 1.3 and 3.6, H-3'), 7.17 (1 H, dd, J 1.3 and 5.5 , H-5'), 6.99 (1 H, dd, 3.6 and 5.5, H-4'), 0.37 (9H, s);
δC (50 MHz; CDC13) 148.2 (C-4, s), 129.7 (C-2', s), 129.3 (C-5, d, JCH 193.8), 127.0 (C-4', d, JCH 171.8), 123.4 (C-5', d, JCH 185.5), 118.9 (C-3', d, JCH 168.8) and -0.76 (q, JCH 120.7);
m/z 195 (19.6%, M-N2), 181 (15.0), 180 (100), 83 (37.5), 73 (34.2), and 43 (14.2) (Found: C, 48.4; H, 5.9; N, 18.5%. C9H13N3SSi requires C, 48.4; H, 5.9; N, 18.8%).
Heat of formation: 80.74 kcal/mol.
Mulliken charges: S, 0.538; C-2', -0.238; С-3', -0.139; C-4', -0.143; C-5', -0.428; N-1, -0.097; N-2, 0.019; N-3, -0.080; C-4, -0.496; C-5, -0.092; Si, 1.373; C(Me), - 0.580.
Total dipole moment: 3.926 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1.709; C2'-C3', 1,390; C3'-C4', 1.428; C4'-C5',1,375; C5'-S, 1.669; C2'-N1, 1,399; N1-N2, 1,366; N2-N3, 1,264; N3-C4, 1,418; C4-C5, 1,410; C5-N1, 1,405; C4-Si, 1,778. Angle (°): S-C2'-C3', 111; C2'-N1-N2, 125; N1-N2-N3, 109; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C2'-C3', 20.
1-(2-Selenophene)-4-(trimethylsilyl)-1,2,3-triazole (2)
(4 days, 83%), m.p. 42-43°C;
vmax/cm-1 3120, 2960, 1250, 845 (SiMe3), 760 and 630;
δH (200 MHz; CDC13) 7.87 (1H, s, H-5), 7.81 (1H, dd, J 1.5 and 5.9, H-5'), 7.29 (1H, dd, J 1.3 and 4.0, H -3'), 7.21 (1 H, dd, J 4.0 and 5.9, H-4), 0.36 (9H, s);
δC (50 MHz; CDC13) 148.1 (C-4, s), 130.9 (C-2', s), 128.6 (C-5', d, JCH 193.6), 128.6 (C-4', d, JCH 176.3 ), 127.8 (C-5, d, JCH 193.0), 119.2 (C-3, d, J JCH 167.2) and -0.70 (9. JCH 119.4);
m/z 243 (18.6%, M-N2), 229 (15.2), 228 (100), 177 (8.1), 138 (10.2),118 (10.1), 93 (10.3), 83 (90.2), 80 (5.8), 73 (98.5), 45 (44.9) and 43 (70.7)
(Found: C, 39.95; H, 4.8; N, 15.5%. C9H13N3SeSi requires C, 40.0; H, 4.85; N, 15.55%).
Heat of formation: 48.34 kcal/mol
Mulliken charges: Se 0.186; C-2', -0.266; C-3, -0.070; C-4', -0.082; C-5’
-0.235; N-1, 0.332; N-2, -0.167; N-3, -0.017; C-4, - 0.305; C-5, -0.247; Si,
0.524; C(Me), - 0.250.
Total dipole moment: 3.399 D
Significant geometric parameters:
Bond lengths (Å): Se-C2', 1.891; C2'-С3', 1.353; C3'-C4', 1.445; C4-C5, 1,345; C5'-Se, 1,892; C2'-N1, 1.408; N1-N2, 1,378; N2-N3, 1,272; N3-C4, 1,406; C4-C5, 1,382, C5-N1, 1,405; C4-Si, 1,828. Angle ("): Se-C2 C3, 111; C2-N1-N2, 127; N1-N2-N3, 107; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 112.
Dihedral angle (°), N2-N1 -C2'-C3', 0.
1-(3-Thiophene)-4-(trimethylsilyl)-1,2,3-triazole (3)
(16 days, 90%), m.p. 96-98°C;
vmax/cm-13120, 2960, 1260 and 855 (SiMe3);
δн (200 MHz; CDC13) 7.93 (1 H, s, H-5), 7.56 (1 H, dd, J 1.7 and 3, H-2), 7.47 (1 H, dd, J 1.7 and 5.2, H-4'), 7.43 (1 H, dd, J 3.0 and 5.2, H-5'), 0.38 (9H, s);
δC (50 MHz; CDC13) 147.6 (C-4, s), 136.6 (C-3', s), 128.3 (C-5, d, JCH 192.4), 127.8 (C-5', d, JCH 189.1), 121.8 (C-4', d, JCH 170.6), 114.7(C2', d, JCH 188.3) and -0.79 (q, JCH 120.0);
m/z 195 (11.8%, M-N2), 181 (14.6), 180 (100), 83 (5.0), 73 (13.0) and 43 (10.9) (Found: C, 48.4; H, 5.9; N, 18.6%).
Heat of formation: 78.91 kcal/mol
Mulliken charges: S, 0.623; C-2', -0.450; С-3', 0.023; C-4, -0.162; C-5', -0.442; N-1, -0.100; N-2, 0.009; N-3, -0.079; C-4, -0.505; C-5, -0.082; Si, 1,374; C(Me), -0.58.
Total dipole moment: 4.076 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1,660; C2'-C3', 1,392; C3'-C4', 1.446; C4'-C5', 1,376; C5'-S, 1.666; C3'-N1, 1.406; N1-N2, 1,365; N2-N3, 1,265; N3-C4, 1,417; C4-C5, 1.404, C5-N1, 1.407; C4-Yes, 1,776.
Angle (°): S-C2'-C3', 111; C2'-C3'-C4', 112; C3'-N1-N2, 125; N1-N2-N3, 109; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C3'-C2', 0.
1-(3-Selenophene)-4-(trimethylsilyl)-1,2,3-triazole (4)
(16 days, 87%), m.p. 72-73°C;
vmax/cm-1 3120-3100, 2960, 1260 and 855 (SiMe3);
δн (200 MHz; CDCl3) 8.14 (1 H, dd, J 1.6 and 2.8, H-2'), 8.11 (1 H, dd, 2.8 and 5.5, H-5'), 7.86 (1 H, s , H-5), 7.78 (1 H, dd, J 1.6 and 5.5, H-4') and 0.36 (9H, s);
δC (50 MHz; CDCl3) 143.4 (C-4, s), 138.1 (C-3', s), 132.4 (C-5", d, JCH 190.7), 128.0 (C-5, d, JCH 191.7), 124.6 (C-4', d, JCH 173.0), 119.2 (C-2', d, JCH 188.7) and -0.67 (q, J 119.4);
m/z 271 (M+, 0.9%), 243 (14.8%, M-N2), 228 (100), 224 (17.0), 200 (7.3), 186 (4.1), 177 (4.1), 138 (6.2), 123 (8.1), 93 (6.3), 84 (26.8), 73 (36.6), 45 (22.4) and 43 (30.2) (Found: C, 40.0; H, 4.85; N, 15.5%).
Heat of formation: 52.19 kcal/mol.
Mulliken charges: Se, 0.199; C-2', -0.229; C-3', -0.091; C-4, -0.102; C-5', -0.231; N-1, 0.304; N-2, -0.1714; N-3, -0.016; C-4, -0.3114; C-5, -0.2393; Si, 0.526; C(Me), -0.25.
Total dipole moment: 4.156 D
Significant geometric parameters
Bond lengths (Å): Se-C2', 1,880; C2'-C3', 1.354; C3'-C4', 1.455; C4'- C5', 1.343; C5'-S, 1.885; C3'-N1, 1,433; N1-N2, 1,380; N2-N3, 1,272; N3-C4, 1,404; C4-C5, 1,383, C4-Si, 1,828. Angle (°): Se-C2'-C3', 109; C2'-C3'-C4', 116; C3'-N1-N2, 124; N1-N2-N3, 108; N3-C4-C5, 106; C4-C5-N1, 105; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C3'-C2', 0.
1-(5-Trimethylsilyl-2-thiophene)-4-trimethylsilyl-1,2,3-triazole (5)
(17 days, 96%), m.p. 67-68°C;
vmax/cm-1 3120, 3110, 3090, 2980, 1260, 840 (SiMe3);
δн (200 MHz; CDCl3) 7.97 (I H, s, H-5), 7.29 (I H, d, J 3.60, H-4), 7.10 (1 H, d, J 3.60, H-3'), 0.36 (9H, s) and 0.33 (9H, s);
δc (50 MHz; CDCl3 CDCl3) 147.6 (C-4, s), 141.5 (C-5', s), 138.1 (C-2', s), 133.4 (C-4', d, JCH 164.7), 128.8 (C-5, d, JCH 194.2,), 119.6 (C-3', d, JCH 167.9), -0.21 (q, JCH 120.8) and -1.1 (q, JCH 118.9);
m/z 267 (40.4%, M-N2), 252 (100), 143 (8.4), 83 (17.9), 73 (75.7), 45 (16.2) and 43 (11.5)
(Found: C, 48.90; H, 7.10; N, 14.20%. Calc. for C12H21N3SSi2: C, 48.80; H, 7.15; N, 14.25%).
Heat of formation: 32.06 kcal/mol.
Mulliken charges: S, 0.695; C-2', -0.256; C-3', -0.191; C-4, -0.109; C-5', -0.759; N-1, -0.095; N-2, -0.002; N-3, -0.071; C-4, -0.506; C-5, -0.073;
Si (Thi), 1,325; C(Me, Thi), -0.570; Si, 1,374; C(Me), -0.570.
Total dipole moment: 3.396 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1,690; C2'-C3', 1,397; C3'-C4', 1,420; C4'-C5', 1,385; C5'-S, 1.656; C2'-N1, 1,399; N1-N2, 1,367; N2-N3, 1,264; N3-C4, 1,417; C4-C5, 1.405, C5-N1, 1.407; C4-Si, 1,777; C5'-Si, 1.788.
Angle (°): S-C2'-C3', 111; C2'-N1-N2, 125; N1-N2-N3, 109; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C2'-C3', 177.
1-(5-Trimethylsilyl-2-selenophene)-4-trimethylsilyl-1,2,3-triazole (6)
(10 days, 96%), m.p. 69-71°C;
vmax/cm-1 2970, 1260 and 845 (SiMe3);
δн (200 MHz; CDCl3) 7.87 (1 H, s, H-5), 7.38 (1 H, d, J 3.8, H-4'), 7.36 (1 H, d, J 3.8, H-3'), 0.37 (9H, s) and 0.34 (9H, s);
δC (50 MHz; CDCl3), N.D. (C-2', 5), 147.9 (C-4, s), 135.0 (C4; d, JCH 164.7), 127.7 (C-5, d, JCH 192.3,), 120.7 (C-3', d, JCH 164.6,), 146.8. (C-5', s), 0.45 (q, JCH 119.3) and -0.85 (q, JCH 121.0);
m/z 343 (0.4%, M+), 315 (21.9%, M-N2), 300 (57.9), 298 (19.8), 73 (100), 45 (18.2) and 43 (12.6)
(Found: C, 42.10; H. 6.15; N, 12.25%, Calc. for C12H21N3SeSi2: C, 42.10; H, 6.20; N, 12.30%).
Heat of formation: -6.69 kcal/mol.
Mulliken charges: Se, 0.229; C-2', -0.288; C-3', -0.117; C-4', -0.046; C-5', -0.343; N-1, 0.347; N-2, -0.171; N-3, -0.016; C-4, -0.316; C-5, -0.242; Si (Sel), 0.489; C(Me, Sel), -0.250; Si, 0.523; C (Me), - 0.250.
Total dipole moment: 4.552 D
Significant geometric parameters
Bond lengths (Å): Se-C2', 1.889; C2'-C3', 1,397; C3'-C4', 1.445; C4'-C5', 1,340; C5'-Se, 1,883; C2'-N1, 1.406; N1-N2, 1,378; N2-N3, 1,273; N3-C4, 1,404; C4-C5, 1,385, C5-N1, 1,401; C4-Si, 1,826; C5'-Si, 1.791.
Angle (°): S-C2'-C3', 111; C2'-N1-N2, 121; N1-N2-N3, 108; N3-C4-C5, 106; C4-C5-N1, 105; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C2'-C3', 180.
1-(5-Methyl-2-thiophene)-4-(trimethylsilyl)-1,2,3-triazole (7)
(17 days at +4 °C, 63%), m.p. 65-66°C;
vmax/cm-1 2960, 2920, 1255 and 845 (SiMe3);
δH (200 MHz; CDCl3) 7.78 (1 H, s, H-5), 6.97 (1 H, d, J 3.7, H-3'), 6.64 (1 H, dq, J 3.7 and 1.1, H-4'), 2.47 (3 H, d, J 1.1) and 0.34 (9H, s);
δC (50 MHz; CDCl3) 153.7 (C-4, s), 129.5 (C-2', s), 128.8 (C-5, d, JCH 193.8,), 124.4 (C-4', d, JCH 168.6), 138.0 (C-5', s), 118.6 (C-3', d, JCH 170.0,), 15.5 (q, JCH 129.3, Ме) and -1.1 (q. JCH 119.0, SiMe3);
m/z 237 (M+, 2.5%), 222 (12%, M-Me), 209 (37.2, M-N2), 194 (100),83 (34.7), 73 (35.4) 45 (9.0) and 43 (12.8)
(Found: C, 50.55, H, 6.40, N, 17.60%. C10H15N3SSi requires: C, 50.60; H, 6.40; N, 17.70%).
Heat of formation: 71.83 kcal/mol.
Mulliken charges: S, 0.637; C-2', -0.253; C-3', -0.174; C-4', -0.148; C-5', -0.385; N-1, -0.094; N-2, -0.002; N-3, -0.071; C-4, -0.506; C-5, -0.074; Me(Thi), -0.150; C(Me, Thi), -0.570; Si, 1,374; C(Me), -0.570.
Total dipole moment: 3,460 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1.697; C2'-C3', 1.394; C3'-C4', 1.422; C4'-C5' 1,383; C5'-S, 1.672; C2'-N1, 1,399; N1-N2, 1,367; N2-N3, 1,264; N3-C4, 1,416; C4-C5, 1.405, C5-N7, 1.407; C4-Si, 1,777; C5'-Me, 1,466.
Angle (°) S-C2'-C3', 111; C2'-N1-N2, 124; N1-N2-N3, 109; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C2'-C3', 179.
1-(5-trifluoroacetyl-2-thiophene)-4-trimethylsilyl 1,2,3-triazole (8C4)
(5 days, 98%) m.p. 158-160°C;
vmax/cm-1 3170, 3130, 3120, 3100, 1705(CO), 1185(CF3), 1150 and 850(SiMe3);
δH (200 MHz; CDCl3) 7.98 (1 H, s, H-5), 7.91 (1H, dq, JHH 4.4 and JHF 1.47, H-4'), 7.38 (1 H, d, JHH 4.4, H-3') and 0.39 (9H,5);
δC (50 MHz; CDCl3) 173.9 (q, JCF 37.3, CO), 149.3 (C-4, s), 148.9 (C-2', s), 136.7 (C-4', q, JCF 3.0, JCH 173.8,), 132.4 (C-5', s), 127.8 (C-5, d, JCH 194.4,), 117.8 (C-3', d, JCH 168.1), 116.5 (q, JCF 290.3, CF3) and -1.0 (q, JCF 119.5, SiMe3);
m/z 304 (1.3%, M-Me), 291 (19.5%, M-N2), 276 (82.4), 149 (10.6), 83 (37.8), 77 (15.1), 73 (100), 45 (16.4) and 43 (20.4)
(Found: C, 41.35; H, 3.80; N, 13.10%. C11H12F3N3OSSi requires C, 41.35; H. 3.80;
N, 13.15%).
Heat of formation: - 100.02 kcal/mol.
Mulliken charges: S, 0.657; C-2', -0.205; C-3', -0.169; C-4', -0.059; C-5', -0.492; N-1, -0.117; N-2, 0.032; N-3, -0.070; C-4, -0.478; C-5, -0.090; C(O), 0.285; O, -0.229; C(F), 0.377; F, -0.15; Si, 1,373; C(Me), -0.57.
Total dipole moment: 3.081 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1.692; C2'-C3', 1,400; C3'-C4', 1.415; C4'-C5', 1,392; C5'-S, 1.681; C2'-N1, 1.395; N1-N2, 1,370; N2-N3, 1,260; N3-C4, 1,422; C4-C5, 1,399, C5-N1, 1,415; C4-Si, 1,785; C5'-CO, 1.444; C-O, 1.229; C-CF, 1,572.
Angle (°): S-C2'-C3', 111; C2'-N1-N2, 123; N1-N2-N3, 109; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C2'-C3', 1.
1-(5-trifluoroacetyl-2-thiophene)-5-trimethylsilyl-1,2,3-triazole (8’C5)
(1.5%), m.p. 98-100°C;
vmax/cm-1 1700(CO), 1180(CF3), 1150 and 850(SiMe3);
δH (200 MHz; CDCl3) 7.95 (1 H, dq, JHH 4.15 and JHF 1.5, H-4), 7.81 (1 H, s, H-4), 7.32 (1 H, d, JHH 4.15, H-3') and 0.34 (9H, 5);
m/z 291 (32.1%, M-N2), 277 (15.9), 276 (97.8), 149(11.1), 86 (13.9), 84 (21.2), 83 (23.8), 77 (11.0), 73 (100), 45 (20.8) and 43 (14.0)
(Found: C, 41.35; H, 3.75; N, 13.15%).
1-(5-acetyl-2-thiophene)-4-trimethylsilyl-1,2,3-triazole (9)
(6 days, 97%) m.p. 202-204°C;
vmax/cm-1 3140, 2980, 1650(CO), 1250 and 845 (SiMe3);
δH (200 MHz; CDC13) 7.90 (1 H, s, H-5), 7.62 (1H, d, J 4.1, H-4'), 7.30 (1 H, d, J 4.1, H-3'), 2.58 (3 H, s, Me) and 0.37 (9H, s, SiMe3);
δC (50 MHz; CDCl3) 190.8 (CO, s), 148.6 (C-4, s), 140.9 (C-2', s), 140.4 (C-5', s), 132.1 (C-4 ', d, JCH 169.7,), 128.0 (C-5, d, JCH 193.6,), 117.9 (C-3', d, JCH 168.1,), 26.6 (q, JCH 128.4, Me) and -0.9 (JCH 120.1, SiMe3);
m/z 237 (35.4%, M-N2), 222 (100), 182 (5.5), 83 (22.8), 73 (52.1) and 55 (5.0)
(Found: C, 49.75; H, 5.70; N, 15.80%. C11H15N3OSSi is required C, 49.80; H, 5.70; N, 15.85%).
Heat of formation: 43.46 kcal/mol
Mulliken charges: S, 0.643; C-2', -0.231; C-3', -0.163; C-4', -0.098; C-5', -0.469; N-1, -0.105; N-2, 0.024; N-3, -0.076; C-4, -0.487; C-5, -0.092; C(O), 0.300; O, -0.299; C(H), -0.259; Si, 1,373; C(Me), -0.57.
Total dipole moment: 1.212 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1.695; C2'-C3', 1,397; C3'-CA', 1.418; C4'-C5', 1,388; C5-S, 1,677; C2'-N1, 1,398; N1-N2, 1,368; N2-N3, 1,263; N3-C4, 1,419; C4-C5, 1.401, C5-N1, 1.412; C4-Si, 1,781; C5-CO, 1,462; C-O, 1,238; C-CH, 1,496.
Angle (°): S-C2'-C3', 111; C2'-N1-N2, 123; N1-N2-N3, 109; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C2'-С3', 3.
1-(5-trifluoroacetyl-2-thiophene)-4-trimethylsilyl 1,2,3-triazole (10)
(5 days, 98%) m.p. 158-160°C;
vmax/cm-1 3170, 3130, 3120, 3100, 1705(CO), 1185(CF3), 1150 and 850(SiMe3);
δH (200 MHz; CDCl3) 7.98 (1 H, s, H-5), 7.91 (1H, dq, JHH 4.4 and JHF 1.47, H-4'), 7.38 (1 H, d, JHH 4.4, H-3') and 0.39 (9H,5);
δC (50 MHz; CDCl3) 173.9 (q, JCF 37.3, CO), 149.3 (C-4, s), 148.9 (C-2', s), 136.7 (C-4', q, JCF 3.0, JCH 173.8,), 132.4 (C-5', s), 127.8 (C-5, d, JCH 194.4,), 117.8 (C-3', d, JCH 168.1), 116.5 (q, JCF 290.3, CF3) and -1.0 (q, JCF 119.5, SiMe3);
m/z 304 (1.3%, M-Me), 291 (19.5%, M-N2), 276 (82.4), 149 (10.6), 83 (37.8), 77 (15.1), 73 (100), 45 (16.4) and 43 (20.4)
(Found: C, 41.35; H, 3.80; N, 13.10%. C11H12F3N3OSSi requires C, 41.35; H. 3.80; N, 13.15%).
Heat of formation: - 100.02 kcal/mol.
Mulliken charges: S, 0.657; C-2', -0.205; C-3', -0.169; C-4', -0.059; C-5', -0.492; N-1, -0.117; N-2, 0.032; N-3, -0.070; C-4, -0.478; C-5, -0.090; C(O), 0.285; O, -0.229; C(F), 0.377; F, -0.15; Si, 1,373; C(Me), -0.57.
Total dipole moment: 3.081 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1.692; C2'-C3', 1,400; C3'-C4', 1.415; C4'-C5', 1,392; C5'-S, 1.681; C2'-N1, 1.395; N1-N2, 1,370; N2-N3, 1,260; N3-C4, 1,422; C4-C5, 1,399, C5-N1, 1,415; C4-Si, 1,785; C5'-CO, 1.444; C-O, 1.229; C-CF, 1,572.
Angle (°): S-C2'-C3', 111; C2'-N1-N2, 123; N1-N2-N3, 109; N3-C4-C5, 106; C4-C5-N1, 104; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-C2'-C3', 1.
1-(4-trifluoroacetyl-3-thiophene)-4-trimethylsilyl 1,2,3-triazole (11)
(12 days, 85%) m.p. 99-100°C;
vmax/cm-1 1720(CO), 1180-1170 (CF3) and 850 (SiMe3);
δH (200 MHz; CDCl3) 8.47 (1H, dq, J 3.4 and JHF 1.65, H-5'), 7.90 (1H, s, H-5), 7.73 (1H, d, J 3.4, H-2') and 0.39 (9H, s);
δC (50 MHz; CDCl3) 174.0 (q, JCF 36.6, CO), 146.6 (C-4, s), 128.9 (C-4', s), 138.9 (C-5', q, J JGF 4.8 e JCH 190.9,), 135.9 (C-3', s), 131.7 (C-5, d, JGH 195.4), 124.9 (C-2', d, JCH 192.4,), 116.3 (q, JHF 290.1, CF3) and -1.0 (s, JCH 120.0, SiMe3)
m/z 304 (0.5%, M-Me), 291(16.8%, M-N2), 276 (47.5), 226 (100), 180 (72.5), 136 (13.9), 82 (10.8), 77 (94.9), 73 (41.5), 67 (12.1), 45 (19.6) and 43 (29.4)
(Found: C, 41.35; H, 3.80; N, 13.10. C11H12F3N3OSSi requires C, 41.35; H, 3.80; N, 13.15%).
Heat of formation: -93.20 kcal/moi.
Mulliken charges: S, 0.773; C-2', -0.522; C-3', 0.125; C-4', -0.190; C-5', -0.450; N-1, -0.129; N-2, -0.005; N-3, -0.059; C-4, -0.519; C-5, -0.051; C(O), 0.287; O ,-0.257; C(F), 0.375; F, -0.15; Si, 1,374; C(Me), -0.57.
Dipole total moment: 3.764 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1,640; C2'-C3', 1.405; C3'-C4', 1.458; C4'-C5', 1,399; C5'-S, 1.629; C3'-N1, 1.409; N1-N2, 1,385; N2-N3, 1.257; N3-C4, 1.417; CA-C5, 1.403, C5-N1, 1.409; C4-Si, 1.778; C4'-CO, 1.448; C-0, 1.231; C-CF, 1,586.
Angle (°): S-C2'-C3', 109; C2'-C3'-N1, 120; N1-N2-N3, 110; N3-C4-C5, 106; C4-C5-N1, 105; C4-N3-N2, 111.
Dihedral angle (°), N2-N1-СЗ'-C2', 0.
1-(2-trifluoroacetyl-3-thiophene)-1,2,3-triazole (12)
(80%), m.p. 105-107°C;
vmax/cm-1 3170, 3100, 2960, 1710 (CO), 1180-1170 (CF3):
δH (200 MHz; CDCl3) 8.28 (IH, d, J 1.3, H-4), 7.97 (IH, d, J 5.2, H-5’), 7.85 (IH, d, J 1.3, H-5) and 7.66 (IH, d, J 5.2, H-4');
δC (50 MHz; CDCl3) 172.7 (q, JFC 38.4, CO), N. D. (C-3', s), N. D. (C-2', 5), 135.7 (C-5’, s), 133.8 (C-4,), 126.7(C-4', s), 126.7(C-5, s) and 116.2(q, JFC 287.9, CF3);
m/z 247 (M+, 26.8%), 219 (72.3, M-N2), 150 (100), 122 (69.1), 110 (37.0), 95 (12.0); 82 (20.2), 78 (13.5), 70 (14.4), 69 (20.2), 52 (8.4) and 45 (46.2):
(Found: M+, 247.00272. C8H4F3N3OS requires M+, 247.00272).
Heat of formation: -49.01 kcal/mol
Mulliken charges: S, 0.720; C-2', -0.503; C-3', 0.097; C-4', -0.017; C-5', -0.434; N-1, -0.119; N-2, 0.011; N-3, -0.067; C-4, -0.216; C-5, -0.113; C(O), 0.296; O, -0.190; C(F), 0.383; F, -0.15.
Total dipole moment: 7.182 D
Significant geometric parameters:
Bond lengths (Å): S-C2', 1.669; C2'-C3', 1.403; C3'-C4', 1,440; C4'-C5', 1,379; C5'-S, 1.652; C3'-N1, 1,401; N1-N2, 1,369; N2-N3, 1,263; N3-C4, 1,471; C4-C5, 1,399, C5-N1, 1,412; C2'-CO, 1.456; C-O, 1.224; C-CF, 1,573.
Angle (°): S-C2'-C3', 111; C2'-C3'-N1, 125; N1-N2-N3, 110; N3-C4-C5, 107; C4-C5-N1, 104; C4-N3-N2, 110.
Dihedral angle (°), N2-N1-C3'-C2', 9.
Mixture of silylated C-4 and C-5 triazoles from the azide 10a
(92%, ratio 5.4 : 1.0, 13 days);
δH (200 MHz; CDC13) 8.21 (1H, s, H-5), 7.96 (1H, d, J 5.4, H-5’), 7.63 (1H, d, J 5.4, H-4’) and 0.39 (9H, s) and 7.86 (1H, s, H-4), 7.49 (1H, d, J 5,45, H-5'), 7.08 (1H, d, J 5.45, H-4) and 0.36 (9H, 5).
Spectroscopic properties
Infrared Spectra
In the literature, IR spectra, generally carried out in solution and in the steam phase, of 1,2,3-triazole and some derivatives are reported and the main absorption bands have been analyzed [34-36].
The infrared spectrum of the compounds synthesized (1-9, 11 and 12) shows next to the CH stretching frequencies in the 3140-2960 cm-1 region, two absorption bands at 1250 and 850 cm-1 characteristics of the trimethylsilyl group. The latter are obviously absent in the spectrum of the proto-desylilate compound 12. It is also possible to observe for heteroaryl triazole systems carrying carbonyl-replacement groups a stretching band at approx. 1700-1650 cm-1 in particular for acetyl derivatives 9 the CO stretching band falls at lower frequencies than that of trifluoroacetyl derivatives (8 and 10, 1l and 12). These compounds, in which the trifluoromethyl group is present, have characteristic absorptions at 1180-1185 cm-1 due to C-F stretching.
Mass Spectra
Analysis of the mass spectra of a wide variety of substituted 1,2,3-triazole derivatives, reported in the literature, indicates that the molecular ion M+ is generally intense and almost half of the observed compounds show an M++1 peak. Subsequent fragmentations are highly dependent on the type of substituent [37-40].
Since the 2-H triazole structure does not undergo the primary loss of molecular nitrogen [M-N2], observed instead for most 1-H triazole isomeric derivatives, the different type of fragmentation provides a simple tool to distinguish the two isomers [41].
Nuclear Magnetic Resonance Spectra
The 1H-NMR spectrum, recorded in several solvents, of 1,2,3-triazole and some of its derivatives is reported in the literature. [42] The protons bound to the C-4 or C-5 carbon atoms appear as singlets at 7.9 ppm in deuterochloroform (CDCl3). In other solvents the values are between 8.10 ppm, in deuteropyridine, and 7.42 ppm, in benzene.
There are examples in the literature relating to C-4 or C-5 substituted derivatives of 1,2,3-triazole, however the general trend shows that for 1-substituted derivatives the proton bound to the C-4 carbon atom generally falls to a higher magnetic field than that bound to C-5. The 1H-NMR spectroscopic data related to our 1-heteroaryl-4-trimethylsilyl-1,2,3-triazoles (1-11), show the H-5 proton of the triazole ring as a singlet with a value between 7.82 ppm (1) and 7.98 ppm (8). It is interesting to note that among the reaction products we have obtained, only the trifluoroacetyl derivatives (8, 10 and 11) have provided small quantities of the C-5 silylated regioisomer. In particular, in the case of the reaction of azide 8 it was possible, by chromatographic separation, to obtain the two pure isomers 8 and 10. In the case of azide 11a, the regioisomer C-5 silylated 11 was isolated as the main product. An attempt instead to separate the silylated regioisomers C-4 and C-5 from the azide 10a reaction mixture gave the proto-desylylated compound 12 as the only product. This result is in agreement with a previous study, conducted on two acetyl regioisomeric derivatives, obtained by reaction of 2-trimethylsilyl-4-methyl-1,2,3-triazole with acetyl chloride. The formation of the two regioisomers was highlighted by the non-equivalence of the two protons: The H4 proton was assigned a value of δ equal to 7.40 ppm while the H-5 proton was assigned an δ equal to 7.98 ppm. The proto-desilylated compound 12 instead shows a typical AB system consisting of two doublets relating to the protons H-4 and H-5 at values of δ 8.28 ppm and 7.85 ppm, respectively. This attribution is in agreement with the assignments reported in the literature, in some cases also supported by experimental NOE spectroscopy, which predict a value of (H-5) for the 1-substituted 1,2,3-triazoles less than (H-4) in CDCl3 [43]. This behavior can be explained in terms of the electron-withdrawing power of the N=N (or δ +) group which determines a lower magnetic field displacement of the H-4 proton compared to the H-5 proton near the nitrogen N-1.
The 1H-NMR spectra of the heteroaryl core of the heteroaryl triazole derivatives 1-4 consist of a typical ABX system formed by three distinct quartets that can be attributed on the basis of the relative JHH coupling constants, while the substituted heteroaryl triazole derivatives 5-9, 11 and 12 are formed by the typical AB system consisting of two doublets.
Empirically it can be assumed that the electron density on the various protons of the thiophene or selenophene ring can be correlated to the chemical shift of the proton produced by the substituent (ΔSCS). The comparative study of the thiophenic and selenophenic systems linked to the triazole ring indicates a substantial isoelectronicity between the two systems. In fact, there was a good linear correlation between the SCS values of l, 3 and 5 vs 2, 4 and 6 with a slope of the line close to the unit (R = 0.99; n = 11 and p 1.017) as shown in Graph. 1: A similar linear correlation was found between the SCS values of the carbon atoms of azidothiophens 1a, 3a and 5a and the corresponding values of azidoselenophens 2a, 4a and 6a (R 0.996; n 12 and m 0.974; Graph. 2) The slope of the straight line near the unit confirms the comparable isoelectronicity of the two heteroaryl systems, as previously noted.
Considering that the chemical displacement values (SCS) induced by the substituent can be used as a test of the variation of the charge density distribution on a particular carbon atom belonging to appropriate classes of molecules, we have related the experimentally determined values of SCS using the simple Hammett equation:
SCS = rσR + c
The corresponding σR values (obtained by spectroscopic comparisons) NMR and recently critically re-evaluated by Hansch-Leo-Taft [44] relative to the different substituents (H, Me, C(O)CH3, C(O)CF3) vs. experimental SCS values relative to C-2' carbon atoms in para position with respect to the replacement group of thiophenyl-triazole derivatives 1, 5 and 7-9 and the corresponding azido thiophene derivatives 1a, 5a and 7a-9a. Good linear correlations were obtained with coefficients of 0.95 and 0.99 (n 5) respectively and with comparable slopes. These results indicate that for the trimethylsilyl replacer there is less convergence in both cases. Therefore, a negative value corresponds to an increase in electron density on the proton under examination.
The analysis of our experimental values of Σ(Δδ), obtained by comparison of the appropriate heteroaryl triazole with the substituted heteroaryl derivative, shows a +1, +M electronic effect for the methyl substituent (-0.22, -0.35) and a strong -1, -M effect for the electron-withdrawing substituents acetyl and trifluoroacetyl (0.11, 0.63 and 0.19, 0.92) in line with the respective Hammett constants (F = 0.01, 0.33, 0.54; R = -0.18, 0.17, 0.26). In the case of the trimethylsilyl substituent, whose F and R values in the literature are 0.01 and -0.08, an intermediate behavior is noted. This result is generally rationalized as a cancellation of the positive inductive effect due to a "(p→d) back-bonding" phenomenon which makes the trimethylsilyl group closer to the behavior of a resonance electron attractor [45].
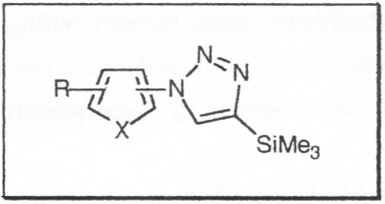
The 13C-NMR spectrum of 1,2,3-triazole was first recorded by Weigert and Roberts [46] in acetone: the chemical shift values corresponding to C-4 and C-5 fall to 130.4 ppm. Subsequently the 13C-NMR spectrum of a number of variously substituted triazole derivatives were reported [11-15]. Analysis of the spectroscopic data indicates that the C-5 carbon atom is generally moved to a higher magnetic field than the C-4 carbon atom. The 13C-NMR spectroscopic data of the 1,2,3-triazole derivatives (1-9, 11 and 12) for solutions at comparable concentrations in CDCl3 are reported in Table 1. The triazole nucleus shows analogies of values referred to the carbon C-5 with those reported in the literature, while the carbon C-4 atom carrying the trimethylsilyl group is moved to a lower magnetic field in a more marked manner compared to the corresponding known values. The known value of the 1JCH coupling constant for 1,2,3-triazoles in CDCl3 is 194.3 Hz [47]- Table 2 shows comparatively the values of 1JCH for a series of derivatives 1,2,3-C-and N-methyl substituted triazoles and for compounds 1-9, 11 which in all cases indicate that the Hz value of 1JCS-HS is slightly greater than the value of JC4-H4. Although the differences are not particularly marked, the placement of the methyl group at the C-4 or C-5 position has been deduced from the relative carbon-proton coupling constants in the respective positions (Table 1 and 2) [48].
Table 1. 13C NMR spectra δC(SCS)a of 1-heteroaryl-4-trimethylsilyl-1,2,3-triazoles 1-9, 11, 12b

Table 2. Coupling constant JCH (Hz) of some cmpds. C and N methyl-substituted and 1-heteroaryl-4-trimethylsilyl-1,2,3-triazoles 1-9, 11
Pharmacology applications of the trimethylsilyl 1,2,3-triazoles
As seen above, 1,2,3-triazoles and their benzo-condensates find wide applications in organic synthesis (e.g. as precursors of azirins and 6-amino-8-azapurines [49,50], in agriculture (as herbicidal, fungicidal and antibacterial pesticides) and in numerous other industrial sectors [11-15]. Our interest is mainly focused on the pharmacological and medical application of triazole derivatives as biologically active systems.
Some 1,2,3-triazole derivatives have shown significant biological activity, e.g. 4-alkyl-1,2,3-triazoles have a mild inhibitory capacity towards microorganisms [51]. In addition, some histamine triazole analogues exhibit antihistamine and anticholinergic activities, while the 1,2,3-triazole ring is an imidazole ring antagonist in the triazole analogues of adenine and guanine [52,53]. The ethanolamine derivatives of 1,2,3-triazoles exhibit mydriatic action [54]. Some biological activities of 1,2,4- and 1,2,3-triazolil [55] are reported in a recent publication with particular regard to the discussion of the virostatic, cytostatic, antimicrobial and antifungal activities of 1,2,3-triazolcarboamide derivatives. In addition, 4- or 5-halomethyl-1,2,3-triazoles, e.g. compound A, can be used as radiomimetics and also show carcinogenic activity [56-58].
.png)
Compound B, 1-tosyl-1,2,3-triazole, possesses bacteriostatic activity [59]. A typical example of an antibiotic that carries a triazole ring in the side chain is cefatrizine, c. This compound belongs to the class of cephalosporins currently in use and is active against 342 different types of germs [60,61]. Another semi-synthetic penicillin, d, derived from a 4-carboxylic acid of 2-phenyl-1,2,3-triazole, has recently been reported in the literature [62]. Benzotriazoles, N-1 and N-2 derivatives, containing glycine and various sugar residues such as polyacetylated B-glucopyranose, B-ribofuranose or furanose were found to be cytostatic [63-65]; in particular, many benzo-replaced benzotriazoles, and, they are cytostatically active against sarcomas and erythroleukomyelosis. In addition, 1H- and 2H-1,2,3-triazole-nucleosides such as G and I possess bactericidal [66] activity, while h and i have been found to be active in vitro against the herpes and measles virus [67,68].
Our research is therefore aimed at studying the potential applicability as biologically active systems (biomodulators) of the triazole derivatives here synthesized.
Screening, which consists of examining the pharmacological activity of the chosen molecules, is still the fundamental way to identify antibiotics and other drugs of both natural and synthetic origin (xenobiotics). The effectiveness of new drugs is generally tested by public or private centers appropriately equipped for laboratory research and statistical data estimate that only one molecule over ten thousand tested reaches clinical use. Specialized American research centers, such as the "Tuberculosis Antimicrobial Acquisition Coordinating Facility" (TAACF) and the "National Cancer Institute" (NCI) performed a primary in vitro screening to determine the antituberculosis, anti-HIV and antitumor activity of the class of C-4 silylated triazoles synthesized by us. In particular, the TAACF integrates the various components of the development process that leads to the discovery of new drugs into a unified system that facilitates the search for potential anti-tuberculous. The facilitation consists of three programs carried out in strictly interdependent Centers or Laboratories; the first is responsible for the acquisition of the compound and the collection and processing of data, the second and third provide the in vitro and in vivo screening service through the "Hansen's Disease Center" and the "Colorado State University" respectively. The entire program is coordinated by the "Southern Research Institute" under the direction of the "National Institute of Allergy and Infectious Diseases" (NIAID).
Bars extending to the right represent the increased sensitivity of the cell line to the tested agent compared to all cell lines tested. Since the scale of the bar is logarithmic, a bar two units to the right implies that the compound provided a response parameter (e.g. GI50) for the cell line at a concentration one hundred times the average concentration required for all cell lines and therefore the cell line is unusually sensitive to that compound. Bars extending to the left correspondingly imply lower-than-average sensitivity. If, for a particular drug and cell line, the desired parameter could not be determined by interpolation, the length of the bar represented is either the highest concentration tested (and the log10 values of the parameters are preceded by the ">" sign) or the lowest concentration tested (and the logio values are preceded by the sign "<"). Values for both limits (> or <) are also calculated at the midpoint used in the graph. All the compounds tested were inactive in the cases under examination, however, some compounds were found to be biologically active (see attached sheets) and showed anti-mold and anti-algae activity.
Cards of TACCF and NCI
Antituberculous activity
The experimental protocol relating to the evaluation of antituberculosis activity in vitro consists of six points:
- Primary screening is conducted at a concentration of 12.5 vg/ml (or molar equivalent of the highest molecular weight compound in a series of congeners) on Mycobacterium tuberculosis H37Rv, using a radiometric system (BACTEC 460). Compounds that exert less than 99% inhibition in the primary assay (MIC > 12.5 ug/ml) are generally no longer evaluated.
- Compounds demonstrating at least 99% inhibitory capacity are retested at lower concentrations on M. tuberculosis H37Rv to determine the minimum inhibitory concentration (MIC) (system CABTEC 460). The MIC is defined as the lowest concentration capable of inhibiting 99% of the inoculum.
- MICs can also be determined in the BACTEC system 460 with nine drug-resistant M. tuberculosis strains; each strain resistant to a single anti-tuberculosis drug.
- The minimum bactericidal concentration (MBC) is then determined by counting the unit-forming colonies of M. tuberculosis H37Rv and the isoniazid-resistant and rifampicin-resistant strains of M. tuberculosis on drug-free media, following exposure to concentrations equivalent to and greater than the MICs previously determined in the respective strain. The MBCs of compounds that are congeners to existing antituberculosis drugs are also determined against the respective resistant strain of M. tuberculosis.
- Simultaneously with the determination of MBC, the cytotoxicity (IC50) of the compounds is tested in VERO cells at concentrations equal to and greater than the MIC for M. tuberculosis H37Rv. After 72 hours of exposure, viability is judged based on the ability of cells to convert methyl tetrazolium (MTT) salts to the product formazan.
- The compounds for which the IC50: MIC ratio is greater than 4 are tested for their ability to inhibit M. tuberculosis H37RV in a monolayer of mouse peritoneal macrophages (EC80: the lowest concentration capable of determining a reduction of 80% in the ability to form colonies in 4 days compared to the control to which the drug was not added) at concentrations twice lower than the IC50 or at concentrations equivalent to 10, 1 and 0.1 the MIC. The postantibiotic effect (PAE) is then determined by the measurement of subsequent growth retardation in drug-free media.
All the compounds examined proved to be inactive; none of the samples were subjected to further study as the results (MIC>12.5 μg/ml, inhib.<50%) are not very significant when compared to rifampicin, one of the first choice drugs in anti-tuberculosis therapy, whose MIC value is decidedly lower and the percentage of inhibition is higher (MIC RMP=0.5 ug /ml, 98% inhibition).
Anti-HIV activity
The products submitted to the NCI were tested as both potential anti-HIV and anticancer agents. The procedure [69] followed for agents active against immunodeficiency virus (HIV) aims to identify compounds that act at any stage of the reproductive cycle of the virus. The test is based on the killing of T4 lymphocytes by HIV. Small quantities of the virus are added to the cells, which die after two cycles of viral reproduction. Agents that interact with virions, cells, or virus gene products to interfere with viral activities will protect the cells from cytolysis. The system is automated in several parts to accommodate a large number of candidate agents and is generally designed to determine anti-HIV activity. However, compounds that degenerate or are rapidly metabolized under culture conditions cannot show their activity in this type of assay. All tests are compared with at least one positive control (e.g. AZT) performed at the same time and under the same experimental conditions. The procedure is divided into five points:
- The candidate agent is dissolved in dimethyl sulfoxide (DMSO, unless otherwise specified) then diluted 1:100 in cell culture medium prior to preparation of serial dilutions on a semi-logarithmic scale. T4 lymphocytes (CEM cell line) are then added and, after a short interval, the HIV-1 virus, resulting in a final dilution of the compound of 1:200. Uninfected cells plus the compound serve as the toxicity control, while infected and uninfected cells lacking the compound serve as the background control.
- The cultures are incubated at 37 °C in a 5% CO2 atmosphere for 6 days.
- The tetrazolium salt, XTT, is added to all the wells and the cultures are incubated to allow the development of the formazano, from which the cellular vitality is determined, with spectrophotometric investigation, and the protective capacity of the test compound.
- Virus-infected cells and treated with the drug are compared with uninfected and drug-treated cells and other appropriate controls (untreated infected cells and untreated uninfected cells, wells containing the drug without the Cells, etc.) in the same plate.
- The data are interpreted by comparison with those of other tests carried out at the same time and therefore the activity of the compound analyzed is determined.
Results of the primary screening relating to the two samples 9 (685427-N/1) and 11 (685428-0/1) taken as an example of the class of C-4 silylated triazoles synthesized by us, are summarized in the sheets reported on pages I and II, respectively. Each sheet is divided into three sections: the sample and test identification section, the section that graphically summarizes the results obtained and the section relating to the tabulated dose-response data.
The second section shows a plot of the log of the concentration of the test sample (such as μg/ml or molar) against the values measured in the test expressed as a percentage of the values of the uninfected and untreated control. The solid line indicates the percentage of survival of HIV-infected cells treated with the sample (at the indicated concentration) in relation to uninfected and untreated controls.
This line expresses the in vitro anti-HIV activity of the tested compound. The dotted line indicates the percentage of surviving uninfected cells treated with the sample compared to uninfected, untreated controls. This line expresses the sample's ability to inhibit cell growth in vitro. And the reference line this particular experiment is indicated by the dotted reference line.
This line shows the degree of cell damage done by the virus in the absence of treatment and is used as a quality control parameter. In this protocol survival values relating to this parameter of less than 50% are considered acceptable. The percentage of protection was calculated from the data and is presented on the left side of the graph. The third section provides a list of the numerical data presented in the graphic section. Furthermore, the approximate values of the 50% effective concentration (EC50) against viral cytopathic effects, the values of the concentration capable of inhibiting cell growth at 50% (IC50) and the therapeutic index (TI = IC50/EC50). All the compounds examined proved to be inactive.
Antitumorali activity
The class of C-4 silylated triazole derivatives, whose anti-HIV activity is tested, were subjected to primary screening for the evaluation of anti-tumor activity. Cellular samples consist of 60 lines in which samples are tested for a minimum of 5 concentrations at logarithmic dilutions. The experimental protocol used consists of continuous exposure to the drug for 48 hours and the sulforhodamine B (SRB) test is used to estimate cell viability or growth capacity: the principle of the test is similar to that commented previously for salts of tetrazolium. The results obtained from the experiments relating to the compounds taken as examples, 9 and 1l, are expressed in the form of a data table, pages III and IV, dose-response curves, pages V and VI, and in a simpler form of "Main Graphs" pages VII and VIII [the calculated measure of effect is percentage growth (PG)]
The data table collects experimental values for different concentrations expressed as log10 (molar or μg/ml) for each cell line. The first two columns describe the type of tumor form (e.g. leukemia, the cell line (e.g. CCRF-CEM) considered. The other two columns list the Mean ODzero and the Mean Octri; the next five columns report the appropriate Mean ODtest for each of the different concentrations. The next five columns list the growth percentages (PG) for each concentration. The GIS response parameters TGI, and LC50 are interpolated values, which represent concentrations at which the PG is +50, 0 and -50, respectively.
Dose-response curves are created by plotting PG values against the log of the corresponding concentration for each cell line. The cell line curves are in turn grouped by type of tumor shape. The horizontal lines provide the PG value at +50, 0 and -50. The concentrations corresponding to the points where the curves intersect these lines are the GI50, TGI and LC50, respectively.
“Mean Graphs” provide a facilitated visual data interpretation of potential selectivity behaviors for particular cell lines or for particular tumor types with respect to the selected response parameter. In fact, this table shows the significant graphs for each of the three main response parameters described above (GI50, TG1 and LC50).
The measured effect of the compound on a cell line is calculated in accordance with the one or the other of the following two expressions:
If (Mean Otest - Mean ODzero) ≥ 0, then
PG = 100 x (Mean ODtest - Mean ODzero)/(Mean ODctrl - Mean ODzero)
If (Mean ODtest - Mean ODzero) < 0, then
PG = 100 x (Mean OD(est - Mean ODzero) / Mean ODzero where:
Mcan OD = The average of the color optical density measurements from the SRB test.
Mean ODzero = just before exposure of the cells to the test compound.
Mcan ODtest = after 48 hours of exposure of the cells to the compound under investigation.
Mean ODctrl = after 48 hours of non-exposure of the cells to the compound under investigation.
The products
were analyzed using appropriate test, from the experimental protocol validated by specialized American research institutes such as the Tuberculosis Antimicrobial Acquisition Facility (TAACF) and the National Cancer Institute (NCI).
REFERENCES
- https://en.wikipedia.org/wiki/Trimethylsilylacetylene.
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. (2002). A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew Chem Int Ed Engl. 41(14):2596-2599.
- Spinelli D, Zanirato P. (1993). On the chemical, NMR, and kinetic properties of 2-azido- and 3-iodothiophene: recent developments. J Chem Soc Perkin 2. 6:1129-1133.
- Coats SJ, Link JS, Gauthier D, Hlasta DJ. (2005). Trimethylsilyl-directed 1,3-dipolar cycloaddition reactions in the solid-phase synthesis of 1,2,3-triazoles. Org Lett. 7(8):1469-1472.
- Chandra AK, Uchimaru T, Nguyen MTJ. (1999). Chem Soc, Perkin. 1:2117.
- Fleming I. (1976). In: Frontier Orbital and Organic Chemical Reactions; Wiley: London.
- Huisgen RJ. (1976). 1, 3-Dipolar cycloadditions. 76. Concerted nature of 1, 3-dipolar cycloadditions and the question of diradical intermediates. Org Chem. 41(3):403-419.
- Halevi EA. (1992). Orbital Symmetry and Mechanism. The OCAMS view; Springer-Verlag: Heidelberg. pp 169-170.
- https://www.google.com/search?client=firefox-b-e&q=TAACAF+Tuberculosis+Antimicrobial+Acquisition
- Salma U, Ahmod S, Zafer Alam Md, Kahn SA. (2024). A review: Synthetic approaches and biological applications of triazole derivatives. Journal of Molecular Structure. 1301(4):137240.
- Aronoff MR, Gold B, Raines RT. (2016). 1,3-Dipolar Cycloadditions of Diazo Compounds in the Presence of Azides. Org Lett. 18(7):1538-1541.
- Vala DP, Vala RM, Patel HM. (2022). Versatile Synthetic Platform for 1,2,3-Triazole Chemistry. ACS Omega. 7(42):36945-36987.
- Bozorov K, Zhao J, Aisa HA. (2019). 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg Med Chem. 27(16):3511-3531.
- Dai J, Tian S, Yang X, Liu Z. (2022). Synthesis methods of 1,2,3-/1,2,4-triazoles: A review. Front Chem. 10:891484.
- Zanirato P. (2009). Synthesis, reactivity and electronic structure of multifarious hetero-aryl and hetero-aroyl azides. ARKIVOC. 2009(1):97-128.
- Professor Paul Herrling was Head of Corporate Research at Novartis. He is also Chairman of the Board of the Novartis Institute for Tropical Diseases.
- Zanirato P. (1991). Reactivity of aryl and heteroaryl azides with vinylsilane and alkynylsilane. Formation of C-silylated 1,2,3-triazolines and 1,2,3-triazoles. J Chem Soc. Perkin Transactions 1. 11:2789-2796.
- Farina L, Zanirato P, Brillante A. (2000). High pressure assisted 1,3-cycloadditions. Synthesis of silylated triazoles from heteroaryl azides. Int J of High Pressure Res. 18(1-6):365-371.
- Foresti E, Di Gioia MT, Nanni D, Zanirato P. (1995). Gazz. Chim. Ital. 125, 151-161 and refs therein.
- Zanirato P. (1995). Structures and supercompiuting at Cineca. Report. pp. 54-160.
- Businell S, Di Martino E, Zanirato P. (2001). New insight Spectroscopic and xray crystallographic data of the resulting silylated 1,2,3-triazoles. ARKIVOC. 2001(1):131-143.
- Bastide J, Henri-Rousseau O. (1978). The chemisry of C-C triple bond, Chapter 11. Cycloadditions and cyclizations involving triple bonds. Book Editor(s): S. Patai.
- Smith PAS. (1984). Aryl and Heteroaryl Azides and Nitrenes In Azides and Nitrenes, Reactivity and Utility. Scriven EFV, Ed. Academic: Orlando. Chap. 3.
- Spagnolo P, Zanirato P. (1978). A convenient synthesis of azidothiophenes and some of their reactions. Org Chem. 43(18):3539-3541.
- Doering WVE, DePuy CH. (1953). Diazocyclopentadiene1. J Am Chem Soc. 75(23):5955-5957.
- Bissel ER, Finger M. (1959). J Org Chem. 24:1256.
- Gronowitz S, Dahlgren K. (1964). Arkiv. Kemi. 21. pp. 201.
- Gronowitz S, Moses P, Hakansson B. (1960). Arkiv. Kemi. 16. pp. 267.
- Davies D, Spagnolo P, Zanirato P. (1995). Synthesis and thermal reactivity of 2-azido-5-trimethylsilyl thiophene and 2-azido-5-methyl thiophene. Perkin Trans. 1(6):613-614.
- Valenti F, Zanirato P. (1999). New insight into experimental and theoretical properties of some para–like substituent of 2-azidothiophenes, Perkin Trans. 2(3):623-627.
- Gronowitz S, Zanirato P. (1994). Preparation, reactivity, NMR properties and semiempirical MNDO/PM3 structural calculations of 2-azido-and 3-azido-selenophene. J Chem Soc Perkin 2. 1994(8):1815-1819.
- Spinelli D, Zanirato P. (1993). On the Chemical, NMR and Kinetic Properties of 2-Azido- and 3-Azidothiophene. J Chem Soc Perkin Trans 2. 1993(6):1129-1133.
- Spagnolo P, Zanirato P. (1988). J Chem Soc Perkin Trans I. pp. 963-976.
- Borello E, Zecchina A. (1962). Ann Chim (Rome). 52:1302.
- Borello E, Zecchina A, Guglielminotti E. (1969). A vibrational assignment for 1,2,3-triazole. J Chem Soc (B). pp. 307-311.
- Stiefvater OL, Jones H, Sheridan J. (1970). Double-resonance-double-search assignment of the microwave spectrum of 1,2,3-triazole. Spectrochim Acta Part A. 26(4):825-833.
- Barlin GB, Batterham TJ. (1967). J Chem Soc B. pp. 516.
- Wamhoff H. (1984). Comprehensive Heterocycl. Chem ed. Katritzky AR, Rees CW, Pots KT, Pergamon Press, Oxford. Vol 5. Parte 4A. pp. 669.
- Gilchrist TL, Gymer GE. (1974). 1,2,3-Triazoles. Adv Heterocycl Chem. 16:33-85.
- Fan WQ, Katritzky AR. (1996). Comprehensive Heterocycl. Chem IInd edn. Katritzky AR, Reese CW, Scriven EFV, Storr RC. Pergamon Press. Vol. 4. p. 1.
- Gilchrist TL, Gymer GE, Rees CW. (1975). Reactive intermediates. Part XXIV. 1H-Azirine intermediates in the pyrolysis of 1H-1,2,3-triazoles. J Chem Soc Perkin Trans 1. pp. 1-8.
- Barlin GB, Batterham TJ. (1967). J Chem Soc B. pp. 516.
- Holzer W. (1991). On the application of NOE difference spectroscopy for spectral and structural assignments with substituted 1,2,3-triazoles. Tetrahedron. 47(47):9783-9792.
- Hansch C, Leo A, Taft RW. (1991). A survey of Hammett substituent constants and resonance and field parameters. Chem Rev. 91(2):165-195.
- Freeburger ME, Spialter L. (1971). Physical organosilicon chemistry. I. Nuclear magnetic resonance studies of o-, m-, and p-substituted phenyltrimethylsilanes. Evidence bearing on the existence of (p.far.d).pi."back-bonding" in phenylsilanes. J Am Chem Soc. 93(8):1894-1898.
- Weigert FJ, Roberts JD. (1968). Nuclear magnetic resonance spectroscopy. Carbon-13 spectra of five-membered aromatic heterocycles. J Am Chem Soc. 90(13):3543-3549.
- Begtrup M. (1976). 13C–H coupling constants as a tool in tautomerism studies of 1,2,3-triazole, 1,2,4-triazole, and tetrazole. J Chem Soc Perkin Trans 2. 1976(6):736-741.
- Rodios NA. (1984). (1984). 13C NMR spectra of 1‐(α‐aroyloxyarylideneamino)‐1,2,3‐triazoles. Identification of 4,5‐unsymmetrically substituted derivatives. Journal of Heterocyclic Chemistry. 21(4):1169-1173.
- Albert A. (1978). In " Nucleic Acid Chemistry". Ed: Townsend LB, Tipson RS. Wiley, NY, USA. Pp. 97.
- Buchardt O. (1976). "Photochemistry of Heterocyclic Compounds". Wiley- Interscience, NY, USA. pp. 403.
- Hartzel W, Benson FR. (1954). Synthesis of 4-Alkyl-V-triazoles from Acetylenic Compounds and Hydrogen Azide1. J Am Chem Soc. 76(3):667-670.
- Sheehan JC, Robinson CA. (1951). The Synthesis of Phenyl-substituted Triazole Analogs of Histamine. J Am Chem Soc. 73(3):1207-1210.
- Sheehan JC, Robinson CA. (1949). J Am Chem Soc. 71:1436.
- Stein ML, D’Antoni L. (1955). Derivati Etanolamminici del Triazolo Vicinale. Science. 10:235-242.
- Bohm R, Karow C. (1981). Biologically active triazoles. Die Pharmazie. 36(4):243-247.
- Contreras A, Sánchez-Pérez RM, Alonso G. (1978). Halomethyl-1,2,3-triazole derivatives: A new type of alkylating agent active in mouse transplantable tumors. Cancer Chemother Pharmacol. 1:243-247.
- Consejo Superior de Investigaciones Cientificas, Span. Pat. 460 433 (1979) (Chem. Abstr., 1979, 91, 193 585).
- Consejo Superior de Investigaciones Cientificas, Span. Pat. 475 496 (1979) (Chem. Abstr., 1979, 91, 107 984).
- Ferrarini PL, Livi O. (1978). 1.3-Dipolar cycloaddition products of 8-azidotetrazol[1,5a][1,8]naphthyridines with alkynes. Farmaco Ed Sci. 33(7):543-550.
- Ueda Y. (1976). Chemotherapy (Tokio). 24:1661.
- Fass RJ. (1978). Comparative in vitro activities of oral cephalosporins and competitive antibiotics against recent clinical isolates. Curr Therap Res. 24(3):352-365.
- Saha U, Das A, Chakraborty S, Gosh M, Roy DK. (1980). 1. Inst Chem. (India). 52:196. (Chem. Abst., 1981, 94, 139 681).
- Ezerskya LY, Petrusha NA. (1971). Onkologiya. 2:45.
- Petrusha NA. (1971). Onkologiya (Kiev). 2:10-12. [Chem. Abstr. 1972, 77, 83533].
- Chernetskii VP, Petrusha NA, Alekseeva JV. (1973). Friziol Aktiv Veshchestra. 5:121.
- ICN Pharmaceuticals, Inc., US. Pat. 3 968 103 (1976) (Chem Abstr. 1976, 85, 94 660).
- ICN Pharmaceuticals, Inc. US. Pat. 3 948 885 (1976) (Chem. Abstr., 1976, 85, 6010).
- Revankar GR, Solan VC, Robins RK, Witkowsky JT. Nucleic Acids Symp. Ser., 1981, 9, 65 (Chem. Abstr., 1982, 96, 35 682).
- Weislow OS, Kiser R, Fine DL, Bader J, Shoemaker RH, Boyd MR. (1989). New soluble-formazan assay for HIV-1 cytopathic effects: application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J Natl Cancer Inst. 81(8):577-586.