Previous Issues Volume 1, Issue 1 - 2016
3 Month Old Female with Prolonged Fever, Pulmonary Nodules, and a Rare Diagnosis of Infantile Takayasu Arteritis: A Multidisciplinary Collaboration
David Leone1, Caitlin Zaner2, Lauren M Cohee3, Jason W Custer4
1University of Illinois at Chicago, Department of Pediatrics (m/c 856), Chicago, IL 60612, USA.
2University of Maryland, Department of Pediatrics Baltimore, MD 21201, USA.
3University of Maryland, Department of Pediatrics, Infectious Disease, 820 Baltimore, MD 21201, USA.
4University of Maryland, Department of Pediatrics, Pediatric Critical Care, 110 S. Paca St., 8th Floor, Ste. 820 Baltimore, MD 21201,USA.
Corresponding Author:David Leone, University of Illinois at Chicago, Department of Pediatrics (m/c 856), 840 S Wood Street (14th floor), Chicago, IL 60612, USA, Tel: 201-919-1759; E-Mail: [email protected]
Received Date: 12 May 2016 Accepted Date: 13 Jun 2016 Published Date: 21 Jun 2016
Copyright © 2016 Leone D
Citation: Leone D, Zaner C, Cohee LM and Custer JW. (2016). 3 Month Old Female with Prolonged Fever, Pulmonary Nodules, and a Rare Diagnosis of Infantile Takayasu Arteritis: A Multidisciplinary Collaboration. Mathews J Pediatr. 1(1): 001.
Background: Takayasu Arteritis is a systemic inflammatory disease that generally affects the great blood vessels. It has predominately been described in the Asian population and primarily affects females, although it ranges all ethnicities and can occur in both males and females. In the pediatric population this condition is exceptionally rare, and is only recently being described more in depth in the literature.
Case Presentation: A three month old Hispanic female presented with three days of high fever, progressive respiratory distress, lethargy, poor feeding, abdominal distension, and a maculopapular rash. Initial laboratory evaluation revealed transaminitis, anemia, and leukocytosis with an elevated immature neutrophil percentage. Infectious disease evaluation was only positive for enterovirus/rhinovirus via polymerase chain reaction from a nasopharyngeal swab. Early chest radiography showed changes consistent with viral pneumonitis. Empiric antibiotics were discontinued after 48 hours after bacterial cultures returned negative; however, the patient continued to have significant respiratory distress. Concerns for Kawasaki vasculitis prompted treatment of intravenous immunoglobulin; however she remained febrile. Computerized tomography of the chest and abdomen ultimately revealed multifocal pulmonary nodules and hepatomegaly. These nodules were later biopsied revealing only inflammatory tissue with no microbial pathogens isolated. The patient continued to have persistent waxing and waning fevers despite antipyretic therapy and required high flow oxygen support for respiratory distress. A repeat echocardiogram was done three weeks into her course that demonstrated giant coronary artery aneurysms. Further magnetic resonance imaging showed large vessel aneurysms and the patients presentation fit all diagnostic criteria for Infantile Takayasu Arteritis.
Conclusions: This case teaches a lesson that when a patients course follows an atypical or unexpected pattern it can be especially important to broaden the differential diagnosis to encompass rarer conditions that may have initially been unsuspected, and to involve other colleagues early as they can approach a case from a different and new perspective. As a result of the combined rationale and expertise of many medical professionals, this case revealed an unusual presentation of an extremely rare disorder, and this highlights the components of a true interdisciplinary success.
Infantile Takayasu Arteritis; Childhood Takayasu Arteritis; Pulmonary Nodules; Coronary Artery Aneurysms; Subclavian Artery Aneurysms; Aortitis; Infliximab.
ABBREVIATIONS PRES: Paediatric Rheumatology European Society; EULAR: European League against Rheumatism; PRINTO: Paediatric Rheumatology International Trials Organization; AHA: American Heart Association; TA: Takayasu Arteritis; i-TA: Infantile Takayasu Arteritis; CSF: Cerebrospinal Fluid; PICU: Pediatric Intensive Care Unit; NICU: Neonatal Intensive Care Unit; CT: Computed Tomography; ESR: Erythrocyte Sedimentation Rate; CRP: C-Reactive Protein; MRI: Magnetic Resonance Imaging; AST: Aspartate Aminotransferase; AST: Alanine Aminotransferase; PCR: Polymerase Chain Reaction; IVIG: Intravenous Immunoglobulin.
AUTHORS CONTRIBUTIONS Drs. Leone and Zaner contributed to the conception and design of this case presentation, performed the literature review, drafted the initial manuscript, and revised the manuscript; Drs. Cohee and Custer contributed to the conception and design of this case presentation and reviewed and revised the manuscript; all authors approved the final manuscript as submitted.
INTRODUCTION
BACKGROUND
Takayasu arteritis is defined as an inflammatory vasculitis that primarily affects the large vessels. Based on the criteria copublished by the Paediatric Rheumatology European Society (PRES), European League against Rheumatism (EULAR) and the Paediatric Rheumatology International Trials Organization (PRINTO), Takayasu Arteritis (TA) requires evidence of abnormalities within the aorta and/or its main branches [1]. Major criteria can be visualized via angiography, Computerized Tomography (CT), or Magnetic Resonance Imaging (MRI) and consist of aneurysms, narrowing/occlusions, dilatations, or wall thickening. These have 100% sensitivity and 99.9% specificity when present [1]. In addition to the major criteria, 1 of 5 minor criteria must be present including: 1) pulse deficits or claudication, 2) four limb blood pressure discrepancy > 10mmHg, 3) bruits or a palpable thrill, 4) hypertension > 95 percentile for age, gender, and height percentile 5) elevated erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) above normal [1]. Takayasu Arteritis rarely occurs in the pediatric population, and often the diagnosis is delayed due to non-specific initial symptoms caused by systemic inflammation [2]. Although the etiology of the disease is unknown, the pathophysiology includes chronic, autoimmune inflammation causing granulomatous changes to the great arteries. This ultimately results in the pathognomonic findings of aneurysms, vascular stenosis, and/or dilatation [3]. The condition is predominantly described in Asian populations, however no ethnicity is spared, and overall females are affected at a rate of 2:1 over males (though it has been reported as high as 8.5:1) [3, 4]. Patients classically present between the 2nd and 4th decade of life [4]. The incidence of Takayasu arteritis in the pediatric population is not known, however it is estimated that annually there may be as many as 2.6 cases per million in North America and 1 case per million in Europe [3, 5].
CASE PRESENTATION
The patient is a three month old Hispanic female who was referred to a community hospital emergency room by her primary care physician with three days of cough, congestion, rhinorrhea, decreased oral intake and urine output, non-bloody/ bilious emesis, fatigue, tachypnea and persistent fever (TMax 39.7° C). Parents described development of a new rash that waxed and waned with each fever. A full sepsis screen was performed including blood, urine and cerebrospinal fluid (CSF) studies; and empiric ceftriaxone and gentamicin were initiated. She was briefly admitted to this hospital for concern of a systemic infectious process and dehydration; however required transfer to a tertiary care center pediatric intensive care unit (PICU) for progressive respiratory distress. Medical history includes premature birth at 34 weeks via caesarean- section, complicated by maternal pre-eclampsia and gestational diabetes. She had a nine day neonatal intensive care unit (NICU) admission for feeding intolerance and hyperbilirubinemia requiring brief phototherapy. Upon discharge she was thriving, growing and developing well. The patient lives with her parents and two healthy siblings. She did not have sick contacts, had not travelled, had no pets, no unusual diet, and no exposure to farm animals or environmental water sources. Her family is Spanish-speaking only, and their history was only significant for a maternal 3rd trimester still-birth of unknown cause. Upon arrival to the PICU, physical exam revealed an irritable and febrile infant (T 38.9° C) in moderate respiratory distress with inspiratory stridor, coarse breath sounds, decreased aeration bilaterally, as well as a prolonged expiratory phase and cough. She was tachycardic with tacky mucus membranes and delayed capillary refill. She was also noted to have rhinorrhea, congestion, mild conjunctival injection, dry erythematous lips, an erythematous hue to her hands and feet, along with painful, erythematous, hyperpigmented papules and plaques on her extremities, chest, abdomen, back and genitals. Her abdomen was soft and mildly distended with a liver edge 3cm below the costal margin. Musculoskeletal exam was significant for edema of the hands and right wrist. Neurologically, she had normal tone and a grossly intact neurological exam without any focal deficits. Initial laboratory testing showed hypernatremia and a mild metabolic acidosis, as well as elevated urine specific gravity and positive urine ketones. Metabolic panel revealed transaminitis of aspartate aminotransferase (AST) 150 units/L, alanine aminotransferase (ALT) 283 units/L and a hypoalbuminemia of 2.5 g/dL. She had a normal ammonia level of 47 μg/ dL, elevated lactate of 2.2 mmol/L, an elevated CRP of 17 mg/ dL, and leukocytosis of 19,000/mm3 with left shift. She also had a normocytic anemia with a hemoglobin of 7.1 g/dL. CSF profile from outside hospital was normal with glucose 65 mg/ dL, protein 45 mg/dL, WBC 2 cells/μL and RBC 560 cells/μL. Blood, urine, and CSF cultures as well as respiratory viral panel were pending at this time. Genetics labs including urine organic acids, plasma amino acids and acylcarnitine profile were obtained. Additional imaging demonstrated a normal abdominal radiograph and an abdominal ultrasound showing an inflamed and edematous gallbladder wall with biliary sludging and hepatosplenomegaly. Echocardiography was obtained at the time of admission, and revealed a patent foramen ovale with left to right shunting, normal biventricular systolic function, and normal coronary arteries. In her early PICU course, the patient required high flow nasal cannula (8L, FiO2 40%) for stabilization of hypoxia (SaO2 88%) and respiratory distress. She continued to remain febrile and was unable to wean off of high flow nasal cannula. Due to anemia she required intermittent transfusions of packed red blood cells was and was nutritionally supported via nasogastric tube feeding. Her initial differential diagnosis included diseases from multiple clinical categories: 1) Infectious (e.g. sepsis from viral or bacterial pathogen with unclear source), 2) Genetics (e.g. inborn error of metabolism, storage disease), 3) Rheumatology (e.g. infantile Kawasaki Disease, neonatal lupus, Juvenile Idiopathic Arthritis), 4) Hematology/Oncology (e.g. infantile malignancy, hemophagocytic lymphohistiocytosis). Pulmonary diseases such as interstitial lung disease or anatomical airway malformations were also being considered due to her respiratory insufficiency. By her fifth day of fever, persistent symptoms and worsening laboratory parameters, it was clear that she was not a typical patient. This was the beginning of what would be a six-week hospital course, which involved interdisciplinary teamwork with thoughtful input from multiple subspecialists.
CLINICAL DECISION MAKING
Given her presentation, an infectious disease work up was prioritized. Blood, urine, and CSF cultures (including fungal and viral) were ultimately negative, and her antibiotic regimen was appropriately tailored and discontinued as final results returned. Her respiratory viral DNA/RNA panel taken at time of admission returned positive for rhinovirus/enterovirus. The fevers remained continuously elevated (greater than 39.5°C). Infectious disease specialists were involved and broadened the differential diagnosis to include disseminated enterovirus as it has been previously described with similar clinical presentations [6, 7]. Stool samples did not contain evidence of enterovirus as tested by polymerase chain reaction (PCR). In continued consultation with specialists, the infectious investigation was expanded over time to include human immunodeficiency virus (HIV), cytomegalovirus (CMV), Epstein-Barr virus (EBV), Bartonella Henslae Parvovirus B19, Herpes Simplex Virus (HSV), Respiratory Syncytial Virus (RSV), Histoplasmosis sp., Legionella pneumophila, Streptococcus pneumonia, Cryptococcus sp. and M. tuberculosis. All testing was negative. As the hospital course evolved Kawasakis Disease was highly considered as a plausible explanation for her persistently high fevers and physical exam findings. Although she did not meet all of the traditional diagnostic criteria of the American Heart Association (AHA), in this age group Incomplete/Atypical Kawasakis Disease can be diagnosed with 3 of the 5 criteria as outlined by the AHA including: fever of = 5 days, cervical lymphadenopathy, changes in extremities (peeling or erythema), polymorphous rash, bilateral conjunctival injection, and oropharyngeal mucosal changes [8]. However, these attenuated criteria can be used for diagnosis only upon visualization of coronary artery pathology, such as dilation or aneurysms. A repeat echocardiogram was performed on day nine of fever due to continued concern for Incomplete Kawasaki Disease. This study was again within normal limits, but it has been shown that coronary artery aneurysms rarely occur prior to ten days after the start of fever [8]. Given the severity of symptoms and clinical suspicion for this ongoing systemic process, she received 2mg/kg of intravenous immunoglobulin (IVIG); however did not receive aspirin because of significant thrombocytopenia at the time. In consultation with rheumatology and cardiology specialists, she received a second dose two days later due to persistently high fevers and elevated acute phase reactants. With this regimen, she experienced an improvement in clinical and laboratory parameters of a systemic inflammatory process. Specifically, she had resolution of fever, improvement of mucus membrane involvement and skin findings, although there still was some mild periungual desquamation noted. These findings alone, however, do not meet the criteria for diagnosis of Kawasaki Disease. Furthermore, she never had any notable lymphadenopathy or thrombocytosis. Unfortunately, this improvement was transient and within 48 hours of IVIG her fevers returned and respiratory symptoms worsened. Upon presentation, the patients bloodwork reflected an unexplained anemia with a low reticulocyte count (0.19%). This failed to improve spontaneously, and she required multiple transfusions in order to avoid symptomatic anemia. In consultation with hematology/oncology specialists, the severity and persistence of her anemia was thought to have been due to a combination of iatrogenic losses through phlebotomy, an age-appropriate transition from fetal to mature hemoglobin, possible iron deficiency, and bone marrow suppression from a virus, inflammatory process, or medication [9]. Dietary iron supplementation was started. On arrival her platelet count was within normal limits; however, over the first week of admission she developed thrombocytopenia (nadir 39,000 plts/μL) with oozing from phlebotomy and arterial line sites. She also had a coagulopathy with a significantly increased partial thromboplastin time (PTT) for age. At this time she had been receiving standard dose heparin for maintenance of an arterial intravenous catheter, and concern was raised for heparin induced thrombocytopenia (HIT). Heparin was discontinued and platelet and fresh frozen plasma transfusions were initiated. Anti-body testing for HIT was ultimately negative. Bone marrow biopsy was not recommended at this time and the thrombocytopenia and anemia eventually normalized. Based on her symptoms including feeding intolerance, severe respiratory distress, metabolic acidosis, hepatosplenomegaly and lack of improvement with IVIG, an inborn error of metabolism was suspected as a rare but possible explanation for her condition. Further, there was a suspicious family history of third trimester fetal loss of unclear cause. Initial ammonia on admission was within normal limits, and serum lactate was slightly elevated. Urine organic acids, plasma amino acids, and an acyl-carnitine profile were sent at admission and eventually returned within normal limits. A work-up for mucopolysaccharidosis - specifically Gauchers disease - was completed, but glucocerebrosidase activity was within normal levels and ultimately ruled out this diagnosis. It has been shown that in addition to vague systemic symptoms, this presentation could be consistent with neonatal lupus specifically causing hepatobiliary disease with resultant cholecystitis, transaminitis, hepatomegaly, and jaundice [10]. Thrombocytopenia has also been shown to occur by maternal circulating auto-antibodies, although this is rare [11]. In addition, there is a wide range of dermatologic manifestations, but these usually occur in sunexposed areas [12]. Finally, this patient did not have any evidence of heart block or other cardiac manifestations that are often associated with, and characteristic of, neonatal lupus [13]. Anti-Rho and Anti-La antibody testing for this child were obtained and returned negative, also ruling out this condition. Throughout her admission the patient was requiring high flow nasal cannula and required a significant amount of support (6-8L at 30-40%). Multiple weaning attempts were made, however she did not tolerate a lower level of flow or oxygen. Serial chest radiographs were obtained and showed no significant changes except for varying degrees of patchy atelectasis that resolved with increased high flow nasal cannula. A high resolution CT scan of the chest of revealed bilateral, multifocal pulmonary nodules of varying sizes in a random distribution ( > 20 nodules, the largest of which measured 1.01 cm in diameter) (See Figure 1).
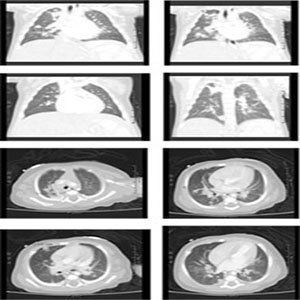
Figure 1: Patients CT Scan showing scattered bilateral, multifocal pulmonary nodules of varying sizes in random distribution throughout all lung fields (> 20 nodules). Largest nodule measures 1.01cm diameter.
It has been reported that pulmonary nodules can arise in Kawasakis Disease; and this was reconsidered given her still persistent fevers and initial constellation of signs and symptoms [14, 15]. A biopsy of a lung nodule was performed via CT guidance. The acquired tissue sample was small and contained large amounts of fibrous tissue. Histopathologically, only a small sample was isolated, which showed only inflammatory cells and connective tissue. Further, it did not grow any fungi, M. tuberculosis, or other bacteria in culture. An open surgical biopsy was planned due to the paucity of tissue and lack of clinical improvement. Repeat echocardiographic imaging was obtained prior to a planned open surgical biopsy to re-evaluate cardiac function and for sequelae of Kawasaki Disease or other vasculitides. Echocardiogram #3 was abnormal with findings including multiple giant coronary artery aneurysms. All specialists were informed, and her surgical excisional biopsy was cancelled. Although coronary artery disease was now identified, it was felt given her persistent illness course with refractory fever despite IVIG, extensive pulmonary involvement, thrombocytopenia, coagulopathy and intra-abdominal findings that she still did not meet accepted criteria for the Incomplete Kawasaki Disease. There was significant concern for more diffuse vascular involvement, so a magnetic resonance angiogram was recommended. This study showed extensive, bilateral and diffuse involvement of the patients coronary, mediumand large-sized arteries. [Please see Figure 2 for detailed description]. According to discussion with multiple rheumatology experts at various institutions and the criteria listed by EULAR, PRINTO, and PRES, these large vessel findings, secured the diagnosis of Infantile Takayasu Arteritis (i-TA)[1].
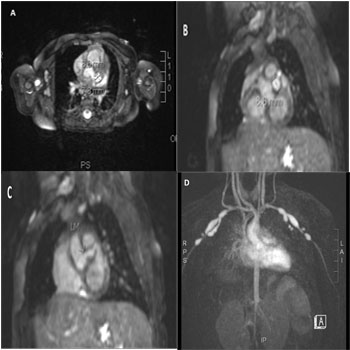
Figure 2: A-C. The largest coronary aneurysms measure 11mm x 8mm x 7mm at the origin of the left anterior circumflex artery, with beaded psuedoaneurysms (up to 8.6mm diameter) of the left main and proximal/ mid left anterior descending artery. D. There are multiple bilateral aneurysms involving the subclavian, axillary and proximal brachial arteries and their branches (the largest on each side measuring 20mm length x 7mm diameter). The common, right and left iliac arteries are attenuated in caliber, with a focal aneurysm of the proximal left common iliac artery. Aortic disease involves narrowing of the suprarenal abdominal aorta (as thin as 2.4mm below the renal arteries) with aortic wall thickening and increased signal enhancement suggestive of aortitis with possible early aneurysm formation.
High dose IV methylprednisolone was administered, along with a one-time dose of infliximab. In addition, lovenox, clopidogrel, and aspirin therapy were initiated to prevent catastrophic thrombosis of the coronary artery aneurysms. Shortly after treatment the patients medical trajectory changed towards recovery. Her respiratory symptoms resolved, as evidenced by ability to steadily wean from high flow nasal cannula to room air within the next week. Other clinical and laboratory markers of systemic illness normalized. Feeding intolerance improved to full oral feeds. Once appropriate, she was transferred from the PICU to the general pediatric ward. She remained stable for the subsequent two days and after over 6 weeks of admission to the hospital, was discharged home with her family.
DISCUSSION
Although TA is generally considered a disease of the great blood vessels, other case reports have demonstrated coronary artery involvement while still meeting the criteria for Takayasu Arteritis. However; similar to this case, there have been multiple reports of i-TA causing both coronary artery stenosis and aneurysms [4, 16, 17]. There are also been reports of coronary involvement in pediatric polyarteritis nodosa and, of course, as the ever feared complication of Kawasaki disease [8, 18- 20]. The diagnosis of Infantile Takayasu Arteritis was given in this case based on the criteria laid forth by three major rheumatalogical societies [1]. In addition, she did not fit all criteria established by the American Heart Association for the diagnosis of Incomplete Kawasaki Disease, and was missing key features such as cervical lymphadenopathy and thrombocytosis [8]. Moreover, she did not sustain a prolonged response to IVIG as is often expected with Incomplete Kawasaki Disease. At this time is in unclear if there is any true difference between Infantile Takayasu Arteritis and Kawasaki Disease or rather a spectrum of inflammatory vasculidites. There are overlapping findings affecting various sized blood vessels in both diseases, and until further investigation is completed on the etiology of each condition, this will only remain as speculation. It should be noted that pulmonary nodules have been described in other vasculitides including Kawasaki Disease [21, 22]. There are few examples in the literature that cite this and only two that investigated the histopathology of pulmonary nodules in Kawasaki Disease; none are published on these findings in Takayasu Arteritis. One highly referenced case series showed via biopsy and pathological staining that these lesions consisted of predominantly mononuclear cells and fibroblastic tissue. In addition, the vascular walls had invasion of IgA plasma cells within the myointima and adventitia [14]. Another review also reported that CD4 T-lymphocytes, B cells, and macrophages are also at fault for some of the vascular abnormalities seen in these conditions [23]. To date there have not been any comparative therapeutic trials for treatment of pediatric TA. This is largely due to the rarity of this disorder. However, there exists a number of treatment protocols used for this condition based on management of other pediatric rheumatologic disorders. Many are targeted at reducing systemic inflammation, and contain a combination of steroids, methotrexate, and more recently, other immunomodulatory medications such as TNF inhibitors [2, 5]. In some severe cases cyclophosphamide has also been utilized with varying affects [2, 5]. More research is needed in order to characterize the safest and most efficacious therapies. This patient responded very rapidly to anti-TNF medication with resolution of systemic inflammatory symptoms. These results have been demonstrated with similar outcomes in other case reports and series [3, 4, 24, 25].
CONCLUSION
Overall, this patient represents an unusual presentation of a rare disease. Diagnosis was initially difficult due to her nonspecific presentation including persistent respiratory insufficiency, systemic inflammatory response syndrome, and feeding intolerance. Atypical mucocutaneous findings, concurrent rhinovirus/enterovirus infection, and pulmonary nodules significantly complicated the clinical picture and delayed diagnosis; as is common with this rare pediatric condition. Through a very thoughtful interdisciplinary approach involving multiple specialty teams, a systematic evaluation was developed that ultimately revealed her rare diagnosis of Infantile Takayasu Arteritis.
ACKNOWLEDGMENTS
The authors would like to thank the patient and her family for their support and consent in writing this case. We also would like to thank our subspecialty consultants for their input and clinical insight.
CONSENT
Informed consent was obtained from the patient and family for publication of this case report and any accompanying images.
REFERENCES
- Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, et al. (2010). EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 69(5), 798-806.
- Szugye HS, Zeft AS and Spalding SJ. (2014). Takayasu Arteritis in the pediatric population: a contemporary United Statesbased single center cohort. Pediatr Rheumatol Online J. 12, 21.
- Mathew AJ, Goel R, Kumar S and Danda D. (2016). Childhood- onset Takayasu arteritis: an update. Int J Rheum Dis. 19(2), 116-126.
- Mohan S, Poff S and Torok KS. (2013). Coronary artery involvement in pediatric Takayasus arteritis: Case report and literature review. Pediatr Rheumatol Online J. 11(4).
- Brunner J, Feldman BM, Tyrrell PN, Kuemmerle-Deschner JB, et al. (2010). Takayasu arteritis in children and adolescents. Rheumatology (Oxford). 49(10), 1806-1814.
- Wolthers KC, Benschop KS, Schinkel J, Molenkamp R, et al. (2008). Human parechoviruses as an important viral cause of sepsislike illness and meningitis in young children. Clin Infect Dis. 47(3), 358-363.
- Hawkes MT and Vaudry W. (2005). Nonpolio enterovirus infection in the neonate and young infant. Paediatr Child Health. 10(7), 383-388.
- Newburger JW, Takahashi M, Gerber MA, Gewitz MH, et al. (2004). Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 110(17), 2747-2771.
- Lee KW, Chow KM, Chan NP, Lo AO, et al. (2009). Piperacillin/ tazobactam Induced Myelosuppression. J Clin Med Res. 1(1), 53-55.
- Lee LA, Sokol RJ and Buyon JP. (2002). Hepatobiliary disease in neonatal lupus: prevalence and clinical characteristics in cases enrolled in a national registry. Pediatrics. 109(1), E11.
- Watson R, Kang JE, May M, Hudak M, et al. (1988). Thrombocytopenia in the neonatal lupus syndrome. Arch Dermatol. 124(4), 560-563.
- Weston WL, Morelli JG and Lee LA. (1999). The clinical spectrum of anti-Ro-positive cutaneous neonatal lupus erythematosus. J Am Acad Dermatol. 40(5 Pt 1), 675-681.
- Buyon JP, Rupel A and Clancy RM. (2004). Neonatal lupus syndromes. Lupus. 13, 705-712.
- Freeman AF, Crawford SE, Finn LS, Lopez-Andreu JA, et al. (2003). Inflammatory pulmonary nodules in Kawasaki disease. Pediatr Pulmonol. 36(2), 102-106.
- Itani MH, Zakhour RG, Haddad MC and Arabi MT. (2010). Prolonged fever with pulmonary nodules in a 4-month-old baby. Pediatr Infect Dis J. 29(8), 784-788.
- Wang EL, Sato Y, Takeichi T and Kitamura O. (2013). Sudden death of an infant with coronary involvement due to Takayasu arteritis. Cardiovasc Pathol. 22(1), 109-111.
- Reddy SM and Reddy SP. (2009). Stenosis of the main stem of the left coronary artery in a teenager with Takayasus arteritis. Cardiol Young. 19(6), 638-640.
- Canares TL, Wahezi DM, Farooqi KM, Pass RH, et al. (2012). Giant coronary artery aneurysms in juvenile polyarteritis nodosa: a case report. Pediatr Rheumatol Online J. 10(1), 1.
- Chamberlain JL III and Perry LW. (1971). Infantile periarteritis nodosa with coronary and brachial aneurysms: A case diagnosed during life. J Pediatr. 78(6), 1039-1042.
- Dodi I, Raggi V, Verna M, Tchana B, et al. (2011). Atipical Kawasaki disease with coronary aneurysm in infant. Ital J Pediatr. 37, 19.
- Akagi K, Abe J, Tanaka K, Tomotaki S, et al. (2015). Kawasaki disease with pulmonary nodules and coronary artery involvement: a report of two cases and a review of the literature. Int J Rheum Dis.
- Manganelli P, Fietta P, Carotti M, Pesci A, et al. (2006). Respiratory system involvement in systemic vasculitides. Clin Exp Rheumatol. 24 (2 Suppl 41), S48-59.
- Johnston SL, Lock RJ and Gompels MM. (2002). Takayasu arteritis: a review. J Clin Pathol. 55(7), 481-486.
- Hoffman GS, Merkel PA, Brasington RD, Lenschow DJ, et al. (2004). Anti-tumor necrosis factor therapy in patients with difficult to treat Takayasu arteritis. Arthritis Rheum. 50(7), 2296-2304.
- Rosenbloom BE and Weinreb NJ. (2013). Gaucher disease: a comprehensive review. Crit Rev Oncog. 18(3), 163-175.