Previous Issues Volume 3, Issue 1 - 2018
Computation of the Diagnostic Uncertainty as an Alternative to the Determination of Measurement Uncertainty for HIV Qualitative Binary Results
Paulo Pereira1*, Sandra Xavier2
1Department of Research, Innovation and Development, Portuguese Institute of Blood and Transplantation, Avenida Miguel Bombarda 6, 1000-208 Lisboa, Portugal.
2Department of Health, Polytechnic Institute of Beja, Beja, Portugal, and Integrated Researcher in NURSE 'IN - Nursing Research Unit for South and Islands.
Corresponding Author: Paulo Pereira, Department of Research, Innovation and Development, Portuguese Institute of Blood and Transplantation, Avenida Miguel Bombarda 6, 1000-208 Lisboa, Portugal, Tel: +351-210063047; E-Mail: paulo.pereira@ipst
Received Date: 17 Sep 2018 Accepted Date: 07 Nov 2018 Published Date: 12 Nov 2018
Copyright © 2018 Pereira P
Citation: Pereira P and Xavier S. (2018). Computation of the Diagnostic Uncertainty as an Alternative to the Determination of Measurement Uncertainty for HIV Qualitative Binary Results. Mathews J HIV AIDS. 3(1): 019
ABSTRACT
Background and objectives: The European Union regulation for Blood Banks does not require the evaluation of measurement uncertainty in virology screening tests. However, its assessment is mandatory for any blood establishment with tests or methods accredited to ISO 15189 specifications. Measurement uncertainty is defined in the VIM and with the methodology principles published in the GUM can be uniquely determined for results expressed as a numerical quantity. Therefore, an alternative approach is required to compute the uncertainty of the binary outcome, recognized as the“diagnostic uncertainty.” This article discusses and proposes alternative (to GUM) models intended for the evaluation of an immunoassay for the screening of anti-HIV-1+2 antibodies and HIV-1 p24 antigen. However, the suggested methodology can be applied to any other test expressing binary results.
Materials and methods: A systematic review of the literature succeeded such as data collection in research for an antiHIV-1+2/HIV-1 p24 immunoassay, and the application of the Bayesian statistics, principally mathematical models of diagnostic accuracy including diagnostic sensitivity, diagnostic specificity, and the related alpha-error, and beta-error. The area under the ROC curve can also be considered on the manufacturing stage or for “in-house” tests.
Results: Se[%] = 100, Se[%] ε[86.2, 100], beta error = 5%; Sp[%] = 99.3, Sp[%] ε [97.4, 99.8], alfa-error = 5%; OA[%] = 98.7%, PA[%] = 95.8%, PA[%] ε [87.9, 100], NA[%] = 98.9%, NA[%] ε [97.8, 99.9]; AUC = 0.99, AUC ε [0.99, 1.00]
Conclusion: The results show that the diagnostic accuracy can be discussed on uncertainty-based thinking. It is demonstrated that each estimator has uncertainty associated, stated by the alpha-error and beta-error to the diagnostic sensitivity and diagnostic specificity for the absolute results, respectively, and the significance level for the 95% CI.
KEYWORDS
Accreditation; Blood Bank; Diagnostic Accuracy; Diagnostic Uncertainty; ISO 15189; Measurement Uncertainty; Risk Assessment; ROC.
INTRODUCTION
In medical laboratories, tests expressing binary results, i.e., positive/negative, present/absent, true/false, are referred to as "qualitative tests." The binary results can be only directly measurable or can be the result of the classification of a numerical quantity result in an ordinal scale for a certain cutoff. The Vocabulary of International Metrology (VIM) defines "measurement uncertainty"as the 'non-negative parameter characterizing the dispersion of the quantity values being attributed to a measurand, based on the information used" (2.26 of [1]). Therefore, it is not intended to qualitative values, metrologically refereed as "nominal property" (1.30 of [1]). ISO/IEC 17025, for the accreditation of testing and calibration tests or methods in laboratories, is published in 1999 [2]. It is the first international standard to require the determination of measurement uncertainty. Later, ISO 15189 purposed specifically for the accreditation of medical laboratory tests or methods considers that the medical laboratory "shall determine the uncertainty of results, where relevant and possible" [3]. The present ISO 15189 edition requires its determination and also recommends that laboratories define the performance requirements for measurement uncertainty and regularly review their estimates [4]. The European Union [5- 9] and the US [10] agencies of Blood Banks do not require its evaluation.
On the risk-based thinking perspective (0.3.3 of [11]), the laboratorian is encouraged to consider "the effect of uncertainty" (3.7.9 of [12]) in quality control phases, such as method validations. Probably, it is somewhat a complex challenge since the technical documents available on measurement uncertainty just include modeling and/or empirical approaches for quantitative values. They do not integrate alternative methodologies to compute the diagnostic uncertainty of the qualitative tests results. Several definitions have been proposing of "diagnostic uncertainty,"differing due to its application. Since the scope of this paper is HIV qualitative tests or methods, let assume the description suggested by Pereira et al. (2015) (13, 5.3.1 of [14]) as 'the risk of false results".[13] also presents an uncertainty vision in the diagnostic accuracy estimates. Nonetheless, the determination of measurement uncertainty is computed in a paper by Pereira et al. (2016) to a screening immunoassay [15]. The standard measurement uncertainty (2.30 of [1] is used to compute the "gray zone" [16], not to evaluate the uncertainty of the binary results.
Although the general metrology recommends using VIM terminology, it is seldom used for virology tests since it does not contain the appropriate vocabulary. Fuentes-Arderiu [17] and Dybkaer [18] have proposed terminologies suitable for qualitative tests, but they remain no well recognized in the med lab. This article adopts the terminology particular to those methods, usually accepted by the Clinical Laboratory and Standards Institute (CLSI) [19].
This paper discusses and suggests a methodology to estimate the diagnostic uncertainty of qualitative results using Bayesian statistics.
METHODS AND MATERIALS
A review of the literature is done. The data collection in research for an anti-HIV-1+2/HIV-1 p24 immunoassay is taken from the Laboratory of Transmissible Agents, Blood and Transplantation Center of Lisbon, Portuguese Institute of Blood and Transplantation (PIBT), Portugal. The human serum or plasma samples are assayed in Prism HIV Ag/Ab Combo (Abbott Diagnostics, Abbott Park, IL, USA) [20]. The assay is an in vitro two-step sandwich chemiluminescent immunoassay for the measurement of the concentration of antibodies to the Human Immunodeficiency Virus (HIV) type 1 (HIV-1) and/or type 2 (HIV-2) and/or HIV p24 antigen. Antibodies and antigen can be detected in serum or plasma of individuals infected with HIV. The Prism HIV Ag/Ab Combo assay uses microparticles coated with recombinant HIV-1/HIV-2 antigen and monoclonal HIV p24 antibody as a solid phase which binds possible HIV antibodies and/or antigen present. After incubation and a washing step, acridinium labeled conjugates, HIV-1 synthetic peptide, and HIV p24 antibodies are added. The occurrence of complexes is determined by addition of an alkaline hydrogen peroxide solution. The chemiluminescent signal is proportional to the level of the anti-HIV-1 and/or anti-HIV-2 antibodies and/or HIV p24 antigen present in the sample. Determining the level of the anti-HIV-1/anti-HIV-2 antibodies and/or HIV p24 antigen is however mostly a screening to categorize a blood donor as HIV-infected or not. The test is calibrated using some plasma samples from subjects not infected with HIV as negative controls and some plasma samples from persons infected with HIV as positive controls. The reagent manufacturer defines a procedure for computing the cutoff value for the number of emitted photons between positives and negatives. This point is analogous to the clinical decision limit.
Bayesian statistic when the diagnosis is known Diagnostic accuracy methods measure the agreement between the binary results and the diagnostic accuracy criteria (i.e., disease/non-disease) (5.3 of [21]). A Bayesian probability framework is adopted. The patients" samples should be carefully selected to prevent "spectrum bias." This phenomenon occurs when the "bias between estimated test performance and true test performance when the sample used for evaluating an assay does not properly represent the entire disease spectrum over the target (intended-use) population" (4.2 of [22]). If the diagnosis is unknown, a comparative method is used to measure the degree of concordance between both binaries (10.2 of [21]).
The measurement of the percentage of true-positive results among the test results for a sample known to be positive for the test is recognized as "diagnostic sensitivity" Se[%] is measured through the mathematical model: [TP/(TP+FN)].100 (eq. 1)
where TP is the number of true-positive results, and FN is the number of false-negative results. On the non-disease sample, the percentage of true-negative results among the test results for a sample known to be negative for the test is known as "diagnostic specificity" Sp[%] which is measured through the model: [TN/(FP+TN)].100 (eq. 2)
where TN is the number of true-negative results, and FP is the number of false-positive results (5.3 of [21]). Transfusion safety requires that Blood Bank immunoassays should have a high sensitivity to ensure that results from infected donors have a high probability to be expressed as 'positive." Thus, the probability of generating false negative results (beta-error) should be minimized at the expense of having a larger probability of generating false positive results (alfa-error). False positives have certainly a negative impact (waste) in the laboratory's budget since initially positive samples are re-tested using more accurate and expensive methods. But this fact is not related to a lack of safety. Table 1 represents a contingency table to measure diagnostic sensitivity and specificity such as the corresponding alfa-error and beta-error.
Table 1: 2x2 Contingency table for diagnostic accuracy.
| Diagnostic accuracy criteria | |||
| Candidate test results | Positive (Disease, D = 1) | Negative (Non-disease, D = 0) | Total |
| Positive (y = 1) | True-positive results (TP) | False-positive results (FP) (alfa-error) | TP + FP |
| Negative (y = 0) | False-negative results (FN) (beta-error) | True-negative results (TN) | FN + TN |
| Total | TP + FN | FP + TN | N |
Let assume a sampling of infected individuals. The diagnosis is based in the clinical diagnosis or positive results from a reference "gold-standard" test. The infected sampling includes specimens with all types and sub-types of Human Immunodeficiency Virus (HIV) with epidemiological prevalence in the geographic area of the PIBT. If the sampling is not representative of the infected population, a bias effect is induced, and the sensitivity could be overestimated. The uninfected sample does not include specimens with known interference factors, including the following: (a) pre-analytical effects such as anti-coagulant, bilirubin, erythrocytes, and hemoglobin; (b) disease effects; and, (c) drug's effect. The interferents can contribute to misclassification of the binary results leading to unrealistic diagnostic accuracy
Theblood donors’ population is the best source for the noninfected sampling since the donors have a long known medical screening record. CLSI EP12-A2 (9.3 of [21]) suggests that test examinations for the diagnostic accuracy evaluation should be performed in reproducibility conditions during 10 to 20 days Low and high limits for the test sensitivity 95% confidence intervals (CI) can be computed respectively from the formulas:
LLse[%]=(Q1,se-Q2,se)/Q3,se·100 (eq. 3)
HLse[%]=(Q1,se+Q2,se)/Q3,se·100 (eq. 4)
where Q1,se=2·TP+1.962 , Q2,se=1.96·(1.962 +4·TP·FN/(TP+FN))½, and Q3,se=2·(TP+FN+1.962 ) (10.1 of [21].
Low and high limits for the test specificity 95% CI are given respectively by: LLsp[%]=(Q1,sp-Q2,sp)/Q3,sp·100 (eq. 5)
HLsp[%]=(Q1,sp+Q2,sp)/Q3,sp·100 (eq. 6)
where Q1,sp=2·TN+1.962 ; Q2,sp=1.96· (1.962 +4·FP·TN/(FP+TN))½, and Q3,sp=2·(FP+TN+1.962 ) (10.1 of [21].
Agreement when the comparator is other than diagnostic accuracy criteria Obviously, if the diagnosis is unknown, the diagnostic accuracy cannot be measured. However, identical models can be used to measure the agreement of test results. Generally, this approach is suggested for "in-house"tests when the diagnosis based on a consensual logic is unavailable. A comparative test is required for this evaluation. The diagnostic accuracy is known, and the sensitivity and specificity must meet the medical laboratory claims.
Table 2 shows a contingency table as the basis for the determination of the agreements. The overall agreement OA[%] is the percentage of the positive and negative results agreement between the two tests and is given by:
Table 2: 2x2 Contingency table for the agreement of binary results.
| Comparative test | |||
| Candidate test results | Positive(x = 1) | Negative (x = 0) | Total |
| Positive (y = 1) | a | b (alfa-error) | a + b |
| Negative (y = 0) | c (beta-error) | d | c + d |
| Total | a + c | b + d | n |
(a+d)/n·100 (eq. 7)
where “a” is the number of candidate test positive results among the positive results in the comparative test, “d” is the number of candidate test negative results among the negative results in the comparative test, and n is the number of samples.
The positive agreement PA[%] is the percentage of the positive results agreement between the two tests and is given by:
a/(a+c)·100 (eq. 10) where “c” is the number of candidate test negative results among the positive results in the comparative test.
Low and high limits for positive agreement 95% CI can be computed, respectively, from the following mathematical models: LLPA[%]=(Q1,PA-Q2,PA)/Q3,PA·100 (eq. 11)
HLPA[%]=(Q1,PA+Q2,PA)/Q3,PA·100 (eq. 12)
where Q1,PA=2·a+1.962 , Q2,PA=1.96·(1.962 +4·a·c·/(a+c))½, and Q3,PA=2·(a+c+1.962 ) (10.2 of [21])..
Finally, the negative agreement NA[%] expresses the percentage of the negative results agreement between the two tests and is given by: d/(b+d).100 (eq. 13) where “b” is the number of candidate test positive results among the negative results in the comparative test.
Low and high limits for negative agreement 95% CI are respectively given by: LLNA[%]=(Q1,NA-Q2,NA)/Q3,nA·100 (eq. 14)
HLNA[%]=(Q1,NA+Q2,NA)/Q3,NA·100 (eq. 15) where Q1,PA=2·d+1.962 , Q2,NA=1.96·(1.962 +4·b·d·/(b+d))½, and Q3,NA=2·(b+d+1.962 ) (10.2 of [21]).. “a”, “b”, “c”, and “d” are respectively equivalent to diagnostic accuracy “true positive”, “false positives”, “false negatives”, and “true negatives.”
RESULTS AND DISCUSSION
A diagnostic accuracy application for diagnostic sensitivity and diagnostic specificity determination is made (nD1 = 24, nD0 = 278). In Blood Bank principally the claimed diagnostic accuracy should be according to what published for a particular method or its generation. Let assume the sensitivity goal as 100%, and the specificity goal of 98%. The 95% CI lower limit should be = 85% for sensitivity and = 90% for specificity . The claimed limits are selected according to the intended use of results. Let assume that the HIV results are used on the validation of blood components. False negative results have a high risk to cause post-transfusion infection. The estimations of the diagnostic accuracy are shown in Table 3. The results met the performance requirements.
Table 3: Results of the diagnostic sensitivity and diagnostic specificity for a screening immunoassay to detect anti-HIV-1+2/HIV-1 antigen,Prism® HIV Ag/Ab Combo (Abbott Diagnostics, Abbott Park, IL, USA).
| 95% CI | |
| Se[%]=24/(24+0)∙100=100 | |
| LLSe[%]=(51.8-3.8)/55.7∙100=86.2 | LLSp[%]=(555.8-6.7)/563.7∙100=97.4 |
| Sp[%]=276/ (2+276)∙100=99.3 | |
| HLSe[%]=(51.8+3.8)/55.7∙100=100 | HLSp[%]=(555.8+6.7)/563.7∙100=99.8 |
Since sensitivity and specificity are equal to 100% and 99.8%, respectively, the absolute results meet the claims. Also, the required CI low limits are reached: 86.2%, and 97.4%, respectively. Since these estimates can be only applied to the samplings, the false negative rate cannot be claimed to be a type of “diagnostic uncertainty.” It is due to the uncertainty to be usually associated with a probability of defect recognized as “risk.” Otherwise, the false negative rate is not a chance but a descriptive statistic.
The 95% CI is interpreted as the interval related to the chance of a person from 95% of the infected population to be classified as positive for sure. On this CI, the beta-error is 0.05 or 5% representing the risk of this null hypothesis to be false. Similarly, on the 95% CI of specificity, the alfa-error is 0.05 or 5% signifying the risk of the alternative hypothesis to be false. Statistically, the CI represents the values for subjects for which the false rates are not statistically significant at the confidence level of 5%
The 95% CI can be interpreted on risk-based thinking as an uncertainty estimator. As the interval amplitude increases, the diagnostic uncertainty directly rises, i.e., the risk to classify false results increases. The interpretation is to some degree similar to what happens with the expanded measurement uncertainty (2.35 of [1]).
The diagnostic accuracy could also be used for the determination of risk components such as false positive results ratio or false negative results ratio. Every time a medical laboratory claims a 95% CI minimum requirement, the number of samples to determine diagnostic accuracy must be considered since it is a limitation of the claim. For example, if the number of patients in the sampling is 29, the low limit for the 95% CI is 88.3%. Consequently claims higher than 88.3% are unrealistic, since the low limit is dependent not only by the number of false results but also by the number of the samples. For a higher claim, the minimum number of samples must be increased.
The study should be reevaluated if significant epidemiological changes are reported. Note that the diagnostic accuracy single results cannot be inferred to the population of the samplings. The inference is only associated with the 95% CI. Also, it must be well known that the inference is to a population with the same epidemiological and biological characteristics of the tested samples. Anyway, it is strongly suggested the use of the 95% CI since it allows a more powerful evaluation of the test performance than the single results.
The drawback of Bayesian probabilities approach is its susceptibility to inappropriate samplings. For example, a small number of samples leads to CI with a relatively large amplitude. This condition decreases the statistical power of the study. Otherwise, incorrectly classified samples in the supposed group of healthy samples induce errors in the specificity estimate. Also, an infected sample that is not representative of the agent prevalence in the population induces a spectrum bias in the estimates.
For the results agreement model, the same samples are used than for the diagnostic accuracy calculus (nPositive = 24, nNegative = 278). Let assume that the overall agreement, positive agreement, and negative agreement shall be = 98%. The overall agreement and negative agreement CI low limit shall be = 80%, and the positive agreement CI low limit shall be to= 85%
Table 4 shows the estimations of results agreement and indicates that the performance requirements have been met. Let emphasize that results agreement of a candidate and a comparative tests pair should be only determined if the diagnosis is unknown. The considerations regarding agreements must be taken with care, since there they can be misunderstood. Occasionally a bad practice is adopted - diagnostic accuracy is computed when sampling with a known diagnosis is not used. The results could be critically biased since the comparator has a poor diagnostic accuracy.
Table 4: Results of the agreement of binary results for a screening immunoassay to detect anti-HIV-1+2/HIV-1 antigen, Prism HIV Ag/Ab Combo (Abbott Diagnostics, Abbott Park, IL, USA).
| 95% CI | |
| OA[%]=(23+275)/302∙100 =98.7 | |
| - | - |
| PA[%]=23/(23+1)∙100=95.8 | |
| LLPA[%]=(49.8-5.7)/55.7∙100=87.9 | HLNA[%]=(553.8- 7.8)/563.7∙100=99.9 |
| NA[%]=275/ (3+275)∙100=98.9 | HLPA[%]=(49.8+5.7)/55.7∙100=100 |
Such as on the diagnostic accuracy estimates, the 95% CI can be interpreted as an uncertainty estimator using the same logic. The performance requirements should follow the same principles than on the diagnostic accuracy model.
Receiver operating characteristic curve/area under the curve The receiver operating characteristic (ROC) curve, and the area under the curve (AUC) are suggested to be applied to determine the best cutoff for a particular test. Usually, it is applied in the manufacturing research and development (R&D)phase or to “in-house” tests in the med lab. The sample concept considered in the diagnostic accuracy is applied to the AUC estimate. The ROC plot shows the entire spectrum of sensitivity-specificity trade-offs per hypothetical cutoff value (discriminators).
The non-parametric Mann-Whitney U statistics compute the AUC also acknowledged as the Mann-Whitney-Wilcoxon test [23], and the concordance measure (4 of [24]) is similar to the trapezoidal model area. Other non-parametric methods such as Bamber [25], Hanley and McNeil method [26], and DeLong [27] models also offer consistent AUC results. While not ignoring the usefulness of this technique, since it can infrequently be practiced in medical laboratory tests, the mathematical models are not discussed further here.
Figure 1 shows the ROC curve for the anti-HIV-1+2/HIV-1 p24 antigen screening test. The samplings are the same than used in the diagnostic accuracy case. The curve suggests that the test is able to predict the true result of the samples accurately. The perfect test point (0, 1.00) is achieved with a cutoff of 1.00
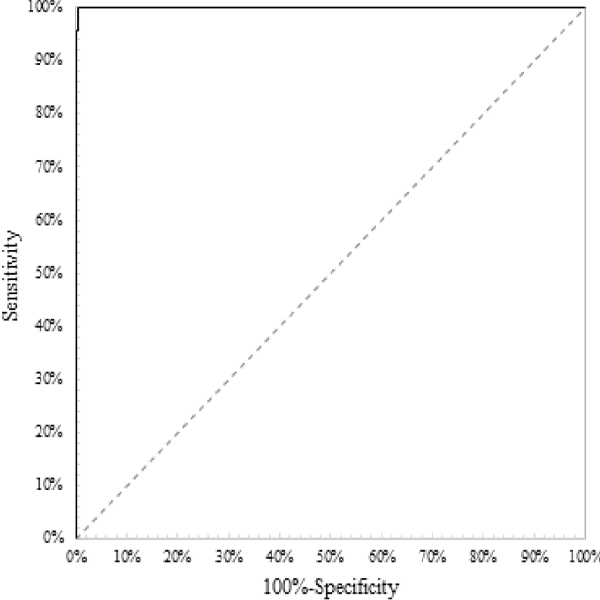
Figure 1: ROC curve for a screening immunoassay to detect anti-HIV-1+2/ HIV-1 antigen, Prism® HIV Ag/Ab Combo (Abbott Diagnostics, Abbott Park, IL, USA) with an AUC = 0.99, AUC ε [0.99, 1.00]
Let assume the next claims: AUC = 0.95, and AUC ε [0.90, 1.00]. AUC of 0.99 is obtained, ranking as an “outstanding discrimination” test [28]. Since the test used is commercial with a known cutoff, the AUC is considered only for demonstration. AUC ε [0.99, 1.00] for what the CI is accepted since the claimed [] contains it. The low limit is close to 1.00 which strengthens the power of the capability to discriminate between true binary results. Identically to the previous evaluations, it is emphasized the use of the 95% CI instead of the single value for a more realistic evaluation of the capacity of a specific test to discriminate true results. Identically to the diagnostic accuracy, if the samplings do not have the same characteristics of the population, the AUC is biased. Once more time, the 95% CI somewhat represents the uncertainty of the AUC. Larger intervals represent a test with a measured weak capability to discriminate results on the inferred population. A con of the AUC con is that it only measures discrimination in the samplings and it is not related uniquely to the infected or non-infected persons.
Moreover, since the sampling of infected individuals is available, the results of the study can be “medically traceable.” In some conditions, the “medical traceability” is difficult to achieve due to the “physicochemical complexity” and “human variability” of patient samples. So, these studies can be not harmonized since there are not available samplings with known diagnosis. For a thorough discussion of traceability in medical laboratories, please refer to [29,30]. Consequently, screening immunoassays for anti-HIV are classified as “methods without traceable calibrators” for what the diagnostic accuracy is untraceable.
All the statistical models are sensitive to outliers. So, the laboratorian should implement a methodology to minimize the risk of misestimation. For instance, use a statistical test such as the Grubbs test [31] when applicable, or alternatively having a second operator revising the results registration.
CONCLUSION
In summary, the diagnostic uncertainty depends on the diagnostic accuracy assessment. The same fact is demonstrated by the agreement of results and the AUC. Just the 95% CI can be viewed on the uncertainty logic. The interpretation is analogous to what happens with the expanded measurement uncertainty. Therefore, the featured models enable the laboratory to evaluate the diagnostic accuracy from a risk-based thinking perspective.
REFERENCES
- Bureau International des Poids et Mesures. (2012). JCGM 200 International vocabulary of metrology - Basic and general concepts and associated terms. 3rd ed. (2008 version with minor corrections).
- International Organization for Standardization. (1999). ISO/IEC 17025 General requirements for the competence of testing and calibration laboratories. Geneva: ISO.
- International Organization for Standardization. (2003). ISO 15189 Medical laboratories - Particular requirements for quality and competence. Geneva: ISO.
- International Organization for Standardization. (2012). ISO 15189 Medical laboratories - Requirements for quality and competence. 3rd ed. Geneva: ISO.
- The European Parliament and the Council of the European Union. (2003). Commission Directive 2002/98/EC Setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components and amending Directive 2001/83/EC. Official Journal of the European Union. 33: 30-40.
- The Commission of the European Communities. (2004). Commission Directive 2004/33/EC Implementing Directive 2002/98/EC of the European Parliament and of the Council as regards certain technical requirements for blood and blood components. Official Journal of the European Union. 91: 25-39.
- The Commission of the European Communities. (2005). Commission Directive 2005/61/EC Implementing Directive 2002/98/EC of the European Parliament and of the Council as regards traceability requirements and notification of serious adverse reactions and events. Official Journal of the European Union. 256: 32-40.
- The Commission of the European Communities. (2005). Commission Directive 2005/62/EC Implementing Directive 2002/98/EC of the European Parliament and of the Council as regards Community standards and specifications relating to a quality system for blood establishments. Official Journal of the European Union. 256: 41- 48.
- European Directorate for the Quality of Medicines. (2018). Guide to the preparation, use and quality assurance of blood components. 19th ed. EDQM. The Council of Europe.
- American Association of Blood Banks (2018). Standards for blood banks and transfusion services. 31st ed. Maryland: AABB.
- International Organization for Standardization (2015). ISO 9001 Quality management systems - Requirements. 5th ed. Geneva: ISO.
- International Organization for Standardization (2015). ISO 9000 Quality management systems - Fundamentals and vocabulary. 3rd ed. Geneva: ISO.
- Pereira P, Westgard J, Encarnaoco P and Seghatchian J. (2015). Evaluation of the measurement uncertainty in screening immunoassays in blood establishments: Computation of diagnostic accuracy models. Transfus Apher Sci. 52(1): 35-41.
- Pereira P. (2016). Measurement uncertainty in screening immunoassays in blood establishments [Ph.D. thesis]. Porto, Portugal: Faculty of Biotechnology, Catholic University of Portugal: 124pp.
- Pereira P, Magnusson B, Theodorsson E, Westgard J, et al. (2015). Measurement uncertainty as a tool for evaluating the 'grey zone' to reduce the false negatives in the immunochemical screening of blood donors for infectious diseases. Accred Qual Assur 21(1): 25-32.
- Kisner HJ. (1998). The gray zone. Clin Lab Manage Rev. 12(4): 277-280.
- Fuentes-Arderiu. (2006). The vocabulary of terms in protometrology. Accred Qual Assur. 11(12): 640-643.
- Dybkaer R. (2007). Metrology and protometrology: the ordinal question. Accred Qual Assur. 12(10): 553-557.
- Clinical Laboratory Standard Institute. (2016). Harmonized terminology database.
- Abbott Diagnostics Division (2008). Abbott Prism HIV Ag/ Ab Combo, code: 77-4254/R6. Wiesbaden: Abbott Laboratories.
- Clinical and Laboratory Standards Institute (2008). EP12- A2 User protocol for evaluation of qualitative test performance. 2nd ed. Wayne (PA): CLSI.
- Clinical and Laboratory Standards Institute (2011). EP24- A2 Assessment of the clinical accuracy of laboratory tests using receiver operating characteristic curves. 2nd ed. Wayne (PA): CLSI.
- Mann H, & Whitney D. (1947). On a test of whether one of two random variables is stochastically larger than the other. Ann Math Statist. 18 (1): 50-60.
- Hastie T, Tibshirani R and Friedman J. (2009). The elements of statistical learning: data mining, inference, and prediction. 2nd ed. New York (NY): Springer.
- Hollander M, Wolfe DA and Chicken E. (2013). Nonparametric statistical methods. 3th ed. New York (NY): John Wiley.
- Bamber D. (1975). The area above the ordinal dominance graph and the area below the receiver operating characteristic curve. J Math Psychol. 12(4): 387- 415.
- Hanley J and McNeil B. (1982). The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 143(1): 29-36
- Swets JA. (1988). Measuring the accuracy of diagnostic systems. Science. 240 (4857): 1285-1293.
- Clinical and Laboratory Standards Institute. (2006). EP32- R Metrological traceability and its implementation, Approved guideline. Wayne (PA): CLSI.
- Vesper H and Thienpont L. (2009). Traceability in laboratory medicine. Clin Chem. 55(6): 1067-1075
- Grubbs F. (1969). Procedures for detecting outlying observations in samples. Technometrics. 11(1): 1-21.